1. INTRODUCCIÓN
El interés terapéutico por el receptor P2X7 comenzó con el descubrimiento de su papel como transmisor de señales de daño en células inmunitarias (1), lo que dispara la activación de procesos inflamatorios. El carácter neuroinflamatorio que se ha otorgado a muchas enfermedades del sistema nervioso central en general (2), y en particular las neurodegenerativas (3), ha disparado el estudio de su fisiología y de la búsqueda de ligandos capaces de modular su acción neurotransmisora. A partir del descubrimiento del papel neurotransmisor del ATP por Burnstock en 1970, se ha descrito la existencia de receptores activados por nucleósidos (receptores purinérgicos P1 activados por adenosina) y nucleótidos (receptores purinérgicos P2) (4), o incluso por bases púricas como adenina (receptores P0) (5), ya en este siglo. Cada uno de estos receptores presenta varios subtipos con diferente funcionalidad y localización. En cuanto a los receptores sensibles a ATP, este es capaz de unirse a receptores metabotrópicos P2Y y a receptores ionotrópicos P2X (6). Los receptores P2X están acoplados a un canal iónico que permite la entrada de iones Na+ y Ca+2 y la salida de K+, aunque además P2X5 es permeable a Cl–. Estos receptores son homo- o heterotrímeros generados por la combinación de siete tipos de subunidades, desde P2X1 a P2X7, si bien es cierto que la subunidad P2X7 solo forma homotrímeros (7). En cualquier caso, las diferencias entre los tipos de receptores P2X se ven reflejadas en su afinidad por ATP, la cinética de activación y cómo se desensibilizan (8) (Figura 1). En concreto, el receptor P2X7, que tiene una cinética de activación relativamente lenta, se diferencia de los demás por su baja afinidad por ATP (EC50 = 0,1 mM) y porque no se desensibiliza (9).
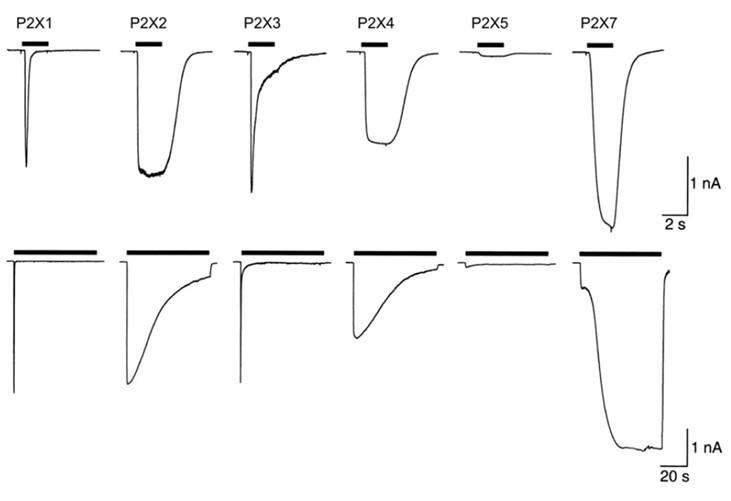
Figura 1. Diferencias en la desensibilización de la corriente para cada subtipo de receptor P2X, en pulsos de 2 s (arriba) o 20 s (abajo) de ATP 30 μM, excepto para P2X7, que fueron de 1 mM. Imagen extraída de Khakh y North, 2012 (8).
Estas características electrofisiológicas le confieren un papel primordial en la propagación de señales de daño y muerte celular por inflamación (piroptosis) (10). El receptor P2X7 está íntimamente en contacto con el inflamasoma NLRP3 (11), de tal manera que la presencia de elevadas concentraciones extracelulares de ATP provoca la activación de P2X7, con la consiguiente salida de K+ por su canal acoplado, señal a la que responde NLRP3 con la activación de caspasa 1 y posterior liberación de citoquinas proinflamatorias (12) (Figura 2). Estas observaciones, entre otras muchas, fundamentan la validación como diana terapéutica en neuroinflamación del receptor P2X7 y justifican el desarrollo de nuevos compuestos que actúen ejerciendo un efecto antagonista potente y selectivo para su posicionamiento en el tratamiento de enfermedades neurodegenerativas (13).
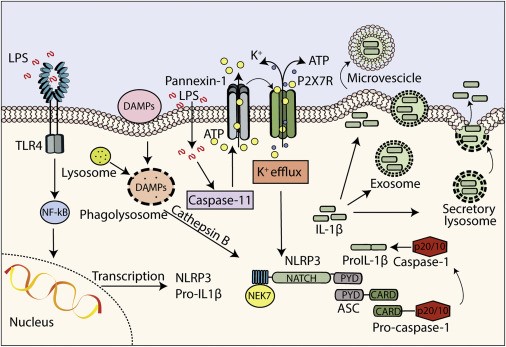
Figura 2. Representación esquemática de la estimulación de la transcripción de interleucina 1β (IL-1β), la activación de NLRP3 inducida por P2X7 y DAMP, y la liberación de IL-1β, como las principales señales inflamatorias asociadas a la activación de P2X7. Imagen tomada de Di Virgilio et al., 2017 (12).
2. CONTEXTO INSTITUCIONAL: LA BÚSQUEDA DE NUEVOS FÁRMACOS DESDE LA UNIVERSIDAD
Nuestro laboratorio está en la Facultad de Ciencias de la Salud de la Universidad Rey Juan Carlos, dentro del Departamento de Ciencias Básicas de la Salud, área de Farmacia y Tecnología Farmacéutica. Esta área, de reciente creación, participa activamente en las tareas académicas del Grado en Farmacia de nuestra Universidad, que empezó su andadura hace 6 años, gracias al tesón y esfuerzo de muchos profesores e investigadores de nuestra facultad, entre los que quiero destacar a la profesora Pilar Carrasco y a la profesora María Jesús Alonso, sin las cuales este grado nunca hubiera cristalizado. Es en este contexto institucional donde se sitúa nuestro grupo de investigación, que aterrizó en la Universidad Rey Juan Carlos hace un lustro, estimulado por este ilusionante proyecto académico. Nuestro interés científico se centra en la I+D de fármacos para el tratamiento de diversas patologías, fundamentalmente neurodegenerativas, que actúan a través de mecanismos singulares o innovadores que han sido poco estudiados, tanto por la industria farmacéutica como por la Academia (14). El abordaje que seguimos sigue las líneas de la Química Farmacéutica, también llamada Médica o Medicinal, y Terapéutica. De forma muy breve, esta disciplina se resume en el estudio de las relaciones estructura-química-actividad biológica. Así, perseguimos la optimización de fármacos para su posible futuro uso clínico a través del entendimiento de las propiedades fisicoquímicas de las moléculas objeto de estudio y cómo estas influyen en la interacción con la diana biológica (receptor, enzima, etc.), sin dejar de lado su papel modulador del perfil farmacocinético y cómo puede influir en su absorción, distribución, metabolismo, excreción y toxicidad en el organismo. El objetivo final es conseguir posicionar nuevos fármacos para el tratamiento de enfermedades. Sin embargo, la tasa de éxito para alcanzarlo es tremendamente baja. Se suele decir que, por cada cien mil compuestos estudiados, como mucho un solo fármaco llegaría a la práctica clínica. Con esta baja eficiencia, cabe preguntarse qué papel puede tener la investigación en I+D de fármacos llevada a cabo en la Academia (Universidades, Organismos Públicos de Investigación, etc.), teniendo en cuenta la limitación de recursos. Esta realidad no ha sido ignorada por nuestro grupo de investigación. Pero dentro de la asunción de que nuestro principal objetivo es el descubrimiento de nuevos fármacos, hemos encontrado un nicho muy fructífero en la obtención de nuevas herramientas farmacológicas útiles para el esclarecimiento del papel de numerosas dianas biológicas en el progreso, agravamiento o mitigación de un proceso patológico, por lo que su uso puede ser de gran interés para grupos de investigación biomédica con los que colaboramos. En suma, nuestra contribución científica ayudaría a la validación de la diana. Incluso podrían ser relevantes en la identificación de nuevas dianas, isoformas o subtipos, tal y como ha ocurrido históricamente en diversas ocasiones, siendo uno de los ejemplos más paradigmáticos el descubrimiento de los diferentes subtipos de receptores adrenérgicos (15).
Con esta idea en mente, en la última década nuestro grupo de investigación ha ayudado a esclarecer el papel que juegan diversas dianas terapéuticas en numerosas enfermedades y a sondear la elegibilidad de estrategias multidiana, tan importantes en el tratamiento de patologías con un carácter multifactorial (16). Así, hemos contribuido a demostrar la relevancia de la activación de la fosfoproteína fosfatasa 2A en la ralentización del progreso de varias enfermedades neurodegenerativas (17), pero también su papel como supresor tumoral (18), o el papel del intercambiador sodio/calcio mitocondrial (19) y el canal CALHM-1 (20) de homeostasia de calcio en la alteración de los niveles de calcio en células excitables y cómo afectan a procesos de muerte/supervivencia neuronal.
No obstante, en los últimos años hemos puesto nuestros esfuerzos en el receptor P2X7 y su papel en la neuroinflamación, que tanto interés despierta en la actualidad. El hecho de que, a diferencia de otros receptores P2X, presente una baja afinidad por ATP, hace que se convierta en un sensor de la presencia de concentraciones elevadas de este, como consecuencia de un escenario de daño celular. Ciertamente, un elevado nivel de ATP extracelular se considera un patrón molecular asociado a daño (DAMP, del inglés “damage associated molecular pattern”) (21).
3. BÚSQUEDA DE ANTAGONISTAS SELECTIVOS P2X7 Y SU RELEVANCIA TERAPÉUTICA
Posterior al reconocimiento del papel de ATP como neurotransmisor y del descubrimiento de sus receptores específicos (P2Y metabotrópicos y P2X ionotrópicos), numerosos trabajos salieron a la luz describiendo ligandos que actuaban como agonistas y antagonistas de estos receptores, con poca selectividad y potencia (22). Estos ligandos eran generalmente fármacos ya conocidos por otras actividades farmacológicas y usos, entre los que destacaban la suramina (23), el derivado de piridoxalfosfato PPADS (24) o KN-62 (25), del que se han preparado interesantes derivados (Figura 3). Entre toda esta 1.ª generación, destaca el tinte Brilliant Blue G (BBG) que, si bien adolece de falta de selectividad, ha sido el estándar usado para el estudio de P2X7 hasta la actualidad (26).
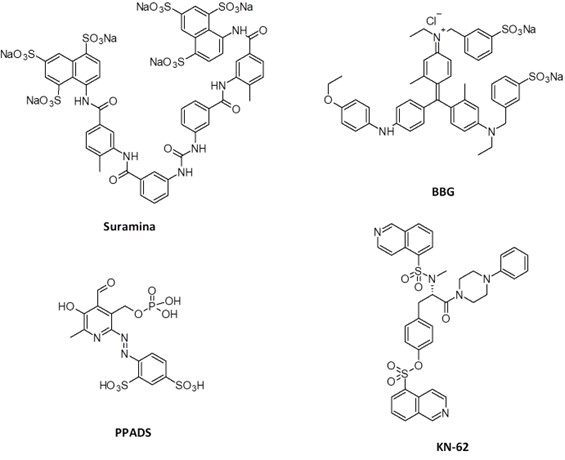
Figura 3. Estructura química de la primera generación de antagonistas de receptores P2X7. BBG, Brilliant Blue G; PPADS, ácido piridoxalfosfato-6-azofenil-2’,4’-disulfonico. Elaboración propia.
Ya en este siglo, la gran industria farmacéutica reconoció la relevancia terapéutica que podría tener el bloqueo selectivo del receptor P2X7, por lo que se embarcó en la búsqueda de nuevas entidades químicas que tuvieran mayor potencia y selectividad, pero que presentaran unas características estructurales que las hicieran elegibles como fármacos de uso clínico. En esta época, encontramos a grandes corporaciones reivindicando familias de fármacos reconocibles para su uso como antagonistas P2X7 (27), como los derivados de adamantano de Astra-Zeneca (28), que han llevado al descubrimiento de AZD9056 (29), el cual llegó a ensayos clínicos en fase II para la artritis reumatoide, guanidinas y tetrazoles de Abbot Laboratories (30, 31), análogos de piroglutámico de GSK (32), las triazinas de Pfizer, cuyo fruto más destacado ha sido CE-224,535 (33), que ha sido estudiado en ensayos clínicos de fase II para la osteoartritis y la artritis reumatoide, o las aportaciones de Jannsen, probablemente la empresa que más empeño ha puesto en el desarrollo de nuevos antagonistas P2X7 (34), destacando su compuesto JNJ-54175446 (35), que se encuentra actualmente en un ensayo clínico en fase II para el trastorno depresivo mayor (36). Podemos decir que JNJ-54175446 es en estos momentos el compuesto de elección utilizado como herramienta farmacológica en la investigación biomédica dedicada a dilucidar el papel fisiológico y patológico de P2X7 (Figura 4).
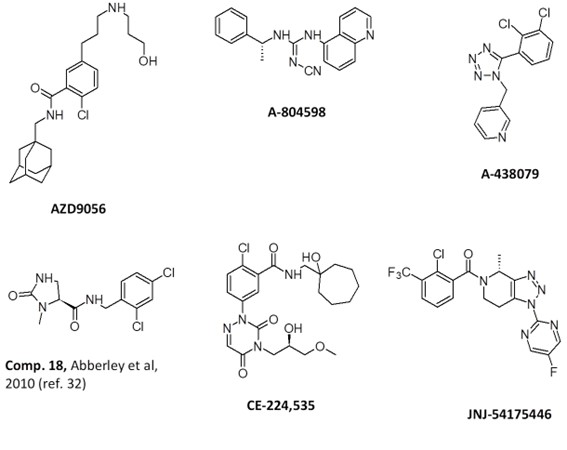
Figura 4. Estructura química de los más destacados cabezas de serie antagonistas de P2X7 desarrollados por la industria farmacéutica (2004 – 2020). Elaboración propia.
4. PURINESDX (2017-2021): ANTAGONISTAS P2X7 PARA ENFERMEDADES DEL SNC
Cuando hace menos de una década nos embarcamos en el ambicioso proyecto de desarrollar nuevos antagonistas P2X7 para su uso en el estudio y tratamiento de enfermedades del sistema nervioso central, dentro de las directrices marcadas por un proyecto financiado por la Unión Europea (PurinesDX, grant agreement #766124, coordinador Prof. Tobias Engel, RCSI, Dublin), lo primero que nos sorprendió fue el vastísimo trabajo realizado para la gran industria farmacéutica, pero a la vez la apreciación de una cierta fatiga, en el sentido de que, después de todo el esfuerzo llevado a cabo, no se habían conseguido los frutos esperados. A finales de la segunda década del siglo XXI, solo la empresa Janssen sigue mostrando un cierto interés. Actualmente, no existe ningún fármaco en clínica que actúe mediante el bloqueo de P2X7, y el único producto en investigación es el JNJ anteriormente mencionado.
Uno cabe preguntarse qué se puede aportar desde la Universidad a este conocimiento, si la industria farmacéutica no ha logrado los hitos deseados, y más aún cuando un proyecto de cuatro años vista exige un suministro rápido y continuo de potenciales compuestos, todo ello con las limitaciones económicas de los fondos públicos. Lo cierto es que esto no nos frenó para comenzar un diseño racional de posibles activos que tuvieran propiedades farmacocinéticas balanceadas, aprovechando además toda la literatura generada en la Academia y la industria. Como se puede apreciar en algunas de las estructuras mostradas en la figura 4, en gran parte de las cabezas de serie y fármacos en clínica desarrollados por la Big Pharma, se encuentra una subestructura de orto-clorobenzamida, unida a diferentes heterociclos nitrogenados. Esta observación dio pie a nuestra hipótesis inicial, que venía a considerar que compuestos que presentaran una estructura esquemática caracterizada por un heterociclo conectado a un anillo aromático halogenado mediante un espaciador de tipo carbonilo o similar, podrían ser un punto de partida estructural para el descubrimiento de posibles antagonistas P2X7. Así, nuestra propuesta de diseño se basó en esta sencilla construcción, donde como heterociclo reclutamos esqueletos de purina, careciendo de la naturaleza nucleotídica que le pudiera generar inespecificidad, recuperando así los primeros intentos de generación de ligandos hechos en la academia hace 30 años, a los cuales se conectarán por un puente carbonílico anillos de benceno altamente halogenados (Figura 5).
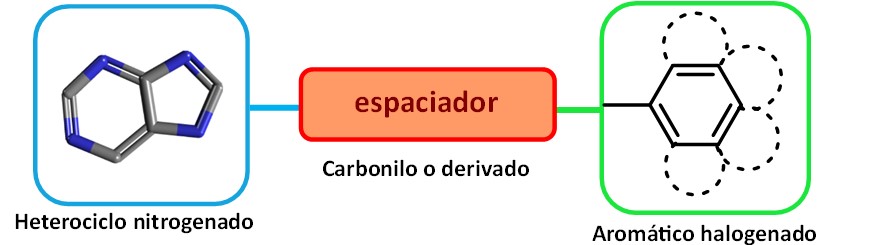
Figura 5. Esquema del diseño sintético para la obtención de nuevos antagonistas P2X7 en nuestro grupo de investigación. Elaboración propia
Sin embargo, lo más importante ha sido tener en cuenta la idiosincrasia propia de un proyecto a corto plazo, donde no solo es que la administración pública exija resultados a los cuatro años, sino que los propios laboratorios del consorcio demandan un suministro de nuevas entidades químicas en los primeros momentos de la ejecución del proyecto, con el fin de sondear el potencial de los nuevos compuestos en las indicaciones terapéuticas que cada colaborador estudiaba. Por esta razón, el diseño químico debía ser lo más sencillo posible, basado en métodos sintéticos fáciles, de alto rendimiento, con purificaciones eficientes y, por encima de todo, escalables, que permitieran la producción de un activo tanto en pocos miligramos como en cantidades multigramos, necesarias en el momento que los miembros del consorcio propusieran el estudio in vivo de los mejores candidatos. No obstante, para proponer qué compuestos podrían evaluar en los modelos de enfermedad, fue necesaria la demostración preliminar de su posible actividad antagonista, razón por la cual nos propusimos poner a punto un modelo in vitro de determinación de la actividad antagonista sobre P2X7 de los derivados obtenidos. En la literatura está bien establecida la captación de la sonda fluorescente YOPRO-1 por cultivos celulares que expresan P2X7 como un método rápido y reproducible, cuando el cultivo se estimula con altas concentraciones de ATP o del agonista más selectivo BzATP (37). La apertura del poro iónico acoplado no solo permite la salida de iones potasio, o la entrada de iones calcio y sodio, sino también la entrada de esta sonda. Así, la reducción de la fluorescencia asociada a la captación de YOPRO-1 permite estimar la potencia antagonista de compuestos, incubados a diferentes concentraciones. Para diseccionar la acción farmacológica, seleccionamos la línea celular HEK293 sobreexpresando de forma estable el receptor P2X7 humano, donado generosamente por Francesco di Virgilio, de la Universidad de Ferrara, Italia, el cual falleció en septiembre de 2024 y a quien expresamos nuestra mayor gratitud por la inestimable ayuda prestada, sin la cual no hubiéramos podido realizar este trabajo.
Ciertamente, la primera familia derivada de N-benzoilpurinas (1, figura 6) no resultó la más satisfactoria, tanto por la falta de actividad como por la elevada inestabilidad química en condiciones suaves. Esta falta de estabilidad suponía un problema grave que debimos acometer con urgencia, por lo que una segunda familia de compuestos fue preparada donde, para conectar purinas y aromáticos halogenados, elegimos el grupo sulfona, de bien conocida estabilidad química y metabólica (2 y 3, Figura 6). Esta serie de compuestos nos ofreció las primeras actividades bloqueadoras de la captación de YOPRO-1. En este punto, es importante mencionar que las urgencias por descubrir un fármaco elegible para su uso en el tratamiento de enfermedades del sistema nervioso central nos obligaron a anticipar los posibles problemas farmacocinéticos que pudieran comprometer su biodisponibilidad central. Es por ello por lo que reclutamos herramientas predictivas computacionales, y sometimos a los compuestos obtenidos a la valoración de las reglas de elegibilidad establecidas por Lipinski hace tres décadas, pero también a las modificaciones propuestas por Hopkins hace unos años, las cuales valoran la posibilidad de que un fármaco tenga un efecto central. Entre los parámetros clave están el valor de log P, que no debía ser menor de 2, y la superficie de área polar, que no debía ser menor de 90 Å2. Lamentablemente, el mejor derivado de la familia 3, al igual que toda la familia de derivados de sulfonamida, fallaron en estos requisitos, lo que nos habla de compuestos muy polares, que probablemente tengan más interés en patologías periféricas. Así, el espaciador tuvo que ser reemplazado, y la siguiente elección residió en una homologación del puente amida, definida por la inserción de un grupo metileno entre el carbonilo y el nitrógeno heterocíclico (4, Figura 6) (38). La primera ventaja encontrada fue la suavidad en las condiciones de obtención, ya que no deja de ser una N-alquilación que se rige por un mecanismo de sustitución nucleófila bimolecular clásico, posible en condiciones suaves en la mayoría de los casos, altos rendimientos y capacidad de escalado mediante técnicas de microflujo.
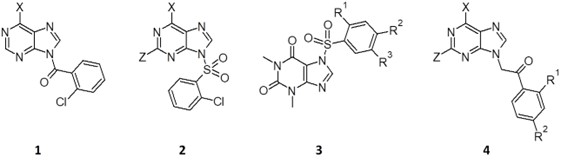
Figura 6. Familias de compuestos desarrollada por nuestro grupo de investigación (38): benzamidas (1), sulfonilpurinas (2), sulfonilteofilinas (3) y acetofenonas (4).
Ciertamente, esta familia de compuestos mostró unos buenos parámetros predictivos fisicoquímicos, y a la hora de la evaluación farmacológica, varios de los compuestos bloquearon la captación de YOPRO-1 en P2X7-HEK293, e incluso mostraron actividad en un segundo ensayo de características más precisas, como es el bloqueo de la corriente purinérgica estimulada por ATP en ovocitos de Xenopus expresando heterologamente P2X7 humano, en una configuración de fijación de voltaje con doble electrodo. De entre todos los derivados, la acetofenona que hemos denominado ITH15004 (Figura 7) se posicionó como el compuesto líder de la familia por ser el más potente, ya que mostró una CI50 de 9 microM en el modelo de YOPRO-1 y redujo la corriente purinérgica en X. laevis casi a la mitad, si bien alcanzó un 75% cuando aumentamos su periodo de incubación hasta 15 min. A destacar es el hecho de que este efecto antagonista se corroboró en X. laevis expresando el receptor P2X7 de rata, pero no en ovocitos transfectados con P2X1, P2X2 y P2X4, lo cual nos habla de su alta selectividad y de su robustez interespecies (38).
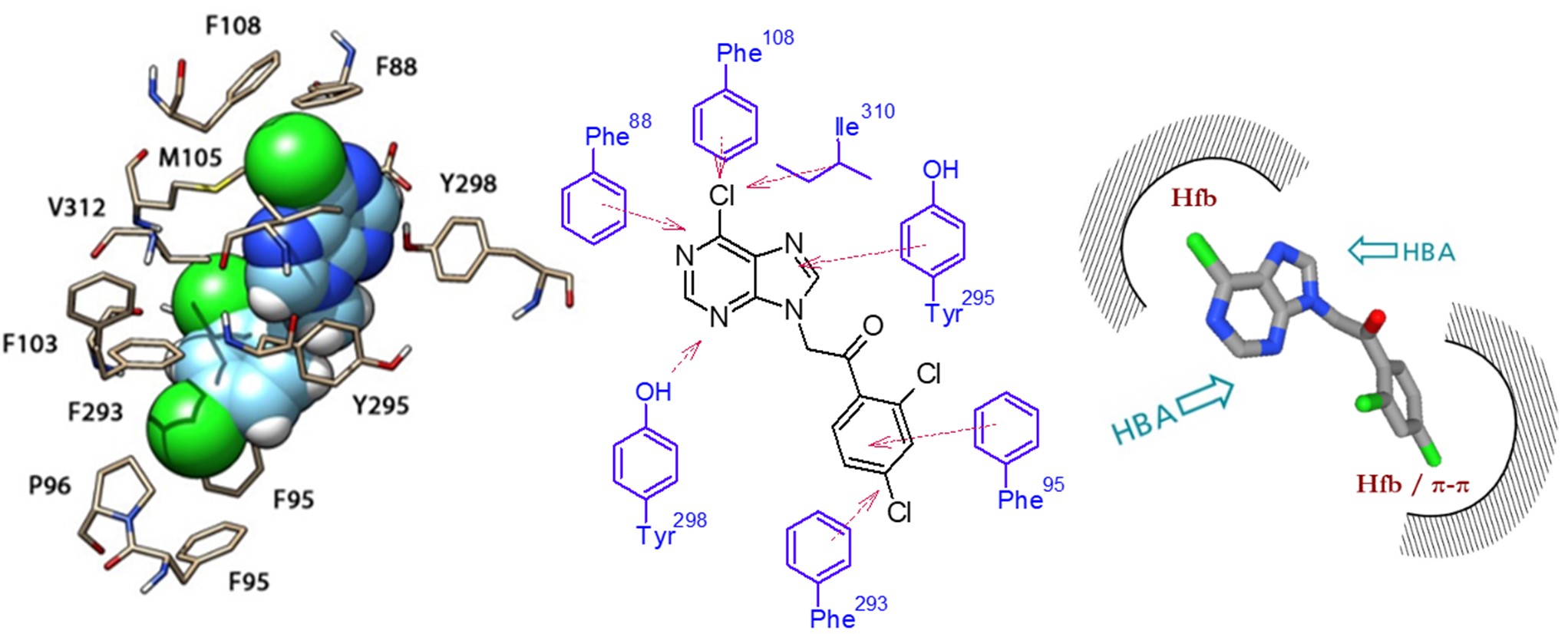
Figura 7. Modo de unión de ITH15004 en el receptor P2X7 (adaptado de Calzaferri et al, 2021, ref. 38), y representación esquemática de las interacciones con los aminoácidos cercanos. Hfb: interacción hidrofóbica. HBA: enlace de hidrógeno como aceptor (Elaboración propia).
Así, ITH15004 es el compuesto que se presentó al consorcio para ser utilizado en el estudio de la influencia de P2X7 en las diferentes enfermedades del sistema nervioso central, gracias además a propiedades farmacocinéticas favorables, experimentales y predictivas, que le convierten en un posible candidato terapéutico: Una buena penetración cerebral (PAMPA), reducida actividad sobre la glucoproteína P, a diferencia del compuesto estrella de Jannsen, y reducidas alertas toxicológicas (predicción con Derek Nexus v6.4.1). Como nota a mejorar, se encuentra su moderada solubilidad acuosa. La reducida solubilidad acuosa de ITH15004 es en parte consecuencia de la hidrofobicidad del sitio de unión con el que interacciona en el receptor. El núcleo de purina se encuentra en contacto con Phe88, Phe108 e Ile310, tres aminoácidos de alto carácter hidrofóbico, al igual que Phe95 y Phe293, que interaccionan con el anillo de fenilo halogenado (Figura 7). Además de estas, encontramos interacciones por enlace de hidrógeno como aceptores a través de los nitrógenos aromáticos del núcleo de purina (38). Futuras optimizaciones tendrán que ir encaminadas a reducir la hidrofobicidad de los ligandos, aprovechando la planaridad de los contactos peptídicos, para lo que favoreceremos interacciones polares por transferencia de carga entre nubes de electrones pi (Figura 7). Estas directrices nos serán de gran ayuda en los próximos años para un nuevo proyecto de investigación que nos han concedido centrado en el desarrollo de nuevos fármacos para el tratamiento de la Esclerosis lateral amiotrófica basado en el antagonismo de P2X7.
Los resultados farmacológicos que hemos obtenido estos últimos años refuerzan nuestra hipótesis, ya que los grupos de investigación biomédica que han utilizado ITH15004 han logrado interesantes frutos. El grupo de investigación de Tobias Engel, del Royal College of Surgeons of Ireland, en Dublín, ha demostrado que la administración de ITH15004 en ratones que sobreexpresan P2X7 sometidos a un modelo de Status epilepticus, se resensibilizan al tratamiento con antiepilépticos, pues la elevada expresión de P2X7 les hacía refractarios a Lorazepam (39). El grupo de Beáta Sperlágh de la Universidad Semmelweis de Budapest demostró que el tratamiento con ITH15004 o con JNJ-54175446 dificulta el buen desarrollo neuronal en estados embrionarios, lo cual subraya la de por sí conocida función fisiológica de P2X7 (40).
Con todo, el compuesto ITH15004 ha despertado el interés de otros grupos de investigación fuera del consorcio PurinesDX en los últimos años. El grupo de investigación del Prof. Manuel Vidal, de la Universidad de Murcia, ha demostrado que ITH15004 puede ser también utilizado para infecciones sistémicas. Ratones sometidos a un modelo de sepsis, sufrieron un daño retiniano que fue mitigado por este antagonista P2X7 solo o en compañía de un antagonista de receptores de TNFalfa (41). Por otra parte, el grupo de investigación del Dr. Javier Egea, interesado en los accidentes cerebrovasculares, ha publicado recientemente los resultados encontrados con varios de los derivados de purina desarrollados por nuestro grupo, encontrando que ITH15004 redujo la liberación de citoquinas proinflamatorias dependiente de P2X7, alivió la piroptosis de cultivos gliales mixtos comprometidos por LPS y ATP, y mejoró las marcas de severidad neurológica en ratones sometidos a un modelo de daño cerebral traumático (42).
6. CONCLUSIONES
En suma, podemos argumentar que hemos comenzado una línea de investigación muy prometedora con los recursos de la Academia, pero asumiendo nuestras limitaciones, tanto de espacios, dinero o tiempos. Los obstáculos presentes los hemos paliado con diversas fortalezas y oportunidades, como es la colaboración con grupos de investigación biomédica especializados. La industria farmacéutica es líder en la I+D de fármacos a través de metodologías de Química Farmacéutica, tanto es así, que en ocasiones desde esta se ha aconsejado a los laboratorios académicos que dejemos de trabajar en esta disciplina tan importante que conjuga Química y Farmacia, y que nos dediquemos únicamente a la identificación de dianas biológicas desde abordajes eminentemente de Biología Molecular. Creemos que la historia que documentamos en esta reseña refuerza la idea de no resignarnos a este rol, y seguir trabajando en la síntesis y evaluación farmacológica de nuevos fármacos, no tanto por entrar en competición con las empresas farmacéuticas, sino por aportar nuestra visión y formar de trabajar. Sin olvidar que es en la Universidad donde se van a formar los futuros profesionales que integrarán las unidades de Química Médica de las diferentes empresas farmacéuticas ubicadas en nuestra Nación.
7. REFERENCIAS
- Burnstock G, Campbell G, Satchell D, Smythe A. Evidence that adenosine triphosphate or a related nucleotide is the transmitter substance released by non-adrenergic inhibitory nerves in the gut. Br J Pharmacol. 1970;40(4):668-88. doi:10.1111/j.1476-5381.1970.tb10646.x.
- Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25(12):1822-32. doi:10.1038/s41591-019-0675-0.
- Adinolfi E, Giuliani AL, De Marchi E, Pegoraro A, Orioli E, Di Virgilio F. The P2X7 receptor: A main player in inflammation. Biochem Pharmacol. 2018;151:234-44. doi:10.1016/j.bcp.2017.12.021.
- Burnstock G. Do some nerve cells release more than one transmitter? Neuroscience 1976;1(4):239-48. doi:10.1016/0306-4522(76)90054-3.
- Bender E, Buist A, Jurzak M, Langlois X, Baggerman G, Verhasselt P, et al. Characterization of an orphan G protein-coupled receptor localized in the dorsal root ganglia reveals adenine as a signaling molecule. Proc Natl Acad Sci. 2002;99(13):8573-8. doi:10.1073/pnas.122016499.
- Burnstock G, Kennedy C. Is there a basis for distinguishing two types of P2-purinoceptor? Gen Pharmacol. 1985;16(5):433-40. doi:10.1016/0306-3623(85)90001-1.
- Kopp R, Krautloher A, Ramírez-Fernández A, Nicke A. P2X7 Interactions and Signaling – Making Head or Tail of It. Front Mol Neurosci. 2019;12:183. doi:10.3389/fnmol.2019.00183.
- Khakh BS, North RA. Neuromodulation by extracellular ATP and P2X receptors in the CNS. Neuron. 2012;76(1):51-69. doi:10.1016/j.neuron.2012.09.024.
- Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic SS. Activation and Regulation of Purinergic P2X Receptor Channels. Pharmacol Rev. 2011;63(3):641-83. doi:10.1124/pr.110.003129.
- Pelegrin P. P2X7 receptor and the NLRP3 inflammasome: Partners in crime. Biochem Pharmacol. 2021;187:114385. doi:10.1016/j.bcp.2020.114385.
- Sperlágh B, Illes P. P2X7 receptor: an emerging target in central nervous system diseases. Trends Pharmacol Sci. 2014;35(10):537-47. doi:10.1016/j.tips.2014.08.002.
- Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity. 2017;47(1):15-31. doi:10.1016/j.immuni.2017.06.020.
- Calzaferri F, Ruiz-Ruiz C, de Diego AMG, de Pascual R, Méndez-López I, Cano-Abad MF, et al. The purinergic P2X7 receptor as a potential drug target to combat neuroinflammation in neurodegenerative diseases. Med Res Rev. 2020;40(6):2427-65. doi:10.1002/med.21710.
- de Los Ríos C, Viejo L, Carretero VJ, Juárez NH, Cruz-Martins N, Hernández-Guijo JM. Promising Molecular Targets in Pharmacological Therapy for Neuronal Damage in Brain Injury. Antioxidants. 2023;12(1):118. doi:10.3390/antiox12010118.
- Kunos G. Adrenergic receptor research: Recent developments. En: Jucker E, editor. Progress in Drug Research [Internet]. Basel: Birkhäuser; 1989 [citado 17 de marzo de 2026]. p. 151-67. doi:10.1007/978-3-0348-9146-2_7.
- de Los Ríos C, Marco-Contelles J. Tacrines for Alzheimer’s disease therapy. III. The PyridoTacrines. Eur J Med Chem. 2019;166:381-9. doi:10.1016/j.ejmech.2019.02.005.
- Arribas RL, Viejo L, Bravo I, Martínez M, Ramos E, Romero A, et al. C-glycosides analogues of the okadaic acid central fragment exert neuroprotection via restoration of PP2A-phosphatase activity: A rational design of potential drugs for Alzheimer’s disease targeting tauopathies. Eur J Med Chem. 2023;251:115245. doi:10.1016/j.ejmech.2023.115245.
- Arribas RL, Bordas A, Domènech Omella J, Cedillo JL, Janssens V, Montiel C, et al. An okadaic acid fragment analogue prevents nicotine-induced resistance to cisplatin by recovering PP2A activity in non-small cell lung cancer cells. Bioorganic Chem. 2020;100:103874. doi:10.1016/j.bioorg.2020.103874.
- Martinez-Sanz FJ, Lajarin-Cuesta R, Gonzalez-Lafuente L, Moreno-Ortega AJ, Punzon E, Cano-Abad MF, et al. Neuroprotective profile of pyridothiazepines with blocking activity of the mitochondrial Na+/Ca2+ exchanger. Eur J Med Chem. 2016;109:114-23. doi:10.1016/j.ejmech.2015.12.043.
- Moreno-Ortega AJ, Martínez-Sanz FJ, Lajarín-Cuesta R, de los Rios C, Cano-Abad MF. Benzothiazepine CGP37157 and its 2′-isopropyl analogue modulate Ca2+ entry through CALHM1. Neuropharmacology. 2015;95:503-10. doi:10.1016/j.neuropharm.2015.02.016.
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805-20. doi:10.1016/j.cell.2010.01.022.
- Murgia M, Hanau S, Pizzo P, Rippa M, Di Virgilio F. Oxidized ATP. An irreversible inhibitor of the macrophage purinergic P2Z receptor. J Biol Chem. 1993;268(11):8199-203.
- Dunn PM, Blakeley AG. Suramin: a reversible P2-purinoceptor antagonist in the mouse vas deferens. Br J Pharmacol. 1988;93(2):243-5. doi:10.1111/j.1476-5381.1988.tb11427.x.
- Lambrecht G, Friebe T, Grimm U, Windscheif U, Bungardt E, Hildebrandt C, et al. PPADS, a novel functionally selective antagonist of P2 purinoceptor-mediated responses. Eur J Pharmacol. 1992;217(2-3):217-9. doi:10.1016/0014-2999(92)90877-7.
- Gargett CE, Wiley JS. The isoquinoline derivative KN-62 a potent antagonist of the P2Z-receptor of human lymphocytes. Br J Pharmacol. 1997;120(8):1483-90. doi:10.1038/sj.bjp.0701081.
- Soltoff SP, McMillian MK, Talamo BR. Coomassie Brilliant Blue G is a more potent antagonist of P2 purinergic responses than Reactive Blue 2 (Cibacron Blue 3GA) in rat parotid acinar cells. Biochem Biophys Res Commun. 1989;165(3):1279-85. doi:10.1016/0006-291X(89)92741-1.
- Bhattacharya A, Biber K. The microglial ATP-gated ion channel P2X7 as a CNS drug target. Glia. 2016;64(10):1772-87. doi:10.1002/glia.23001.
- Guile SD, Alcaraz L, Birkinshaw TN, Bowers KC, Ebden MR, Furber M, et al. Antagonists of the P2X(7) receptor. From lead identification to drug development. J Med Chem. 2009;52(10):3123-41. doi:10.1021/jm801528x.
- Keystone EC, Wang MM, Layton M, Hollis S, McInnes IB, D1520C00001 Study Team. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann Rheum Dis. 2012;71(10):1630-5. doi:10.1136/annrheumdis-2011-143578.
- Donnelly-Roberts DL, Namovic MT, Surber B, Vaidyanathan SX, Perez-Medrano A, Wang Y, et al. [3H]A-804598 ([3H]2-cyano-1-[(1S)-1-phenylethyl]-3-quinolin-5-ylguanidine) is a novel, potent, and selective antagonist radioligand for P2X7 receptors. Neuropharmacology. 2009;56(1):223-9. doi:10.1016/j.neuropharm.2008.06.012.
- Nelson DW, Gregg RJ, Kort ME, Perez-Medrano A, Voight EA, Wang Y, et al. Structure-activity relationship studies on a series of novel, substituted 1-benzyl-5-phenyltetrazole P2X7 antagonists. J Med Chem. 2006;49(12):3659-66. doi:10.1021/jm051202e.
- Abberley L, Bebius A, Beswick PJ, Billinton A, Collis KL, Dean DK, et al. Identification of 2-oxo-N-(phenylmethyl)-4-imidazolidinecarboxamide antagonists of the P2X7 receptor. Bioorg Med Chem Lett. 2010;20(22):6370-4. doi:10.1016/j.bmcl.2010.09.101.
- Duplantier AJ, Dombroski MA, Subramanyam C, Beaulieu AM, Chang SP, Gabel CA, et al. Optimization of the physicochemical and pharmacokinetic attributes in a 6-azauracil series of P2X7 receptor antagonists leading to the discovery of the clinical candidate CE-224,535. Bioorg Med Chem Lett. 2011;21(12):3708-11. doi:10.1016/j.bmcl.2011.04.077.
- Letavic MA, Lord B, Bischoff F, Hawryluk NA, Pieters S, Rech JC, et al. Synthesis and Pharmacological Characterization of Two Novel, Brain Penetrating P2X7 Antagonists. ACS Med Chem Lett. 2013;4(4):419-22. doi:10.1021/ml400040v.
- Chrovian CC, Soyode-Johnson A, Peterson AA, Gelin CF, Deng X, Dvorak CA, et al. A Dipolar Cycloaddition Reaction To Access 6-Methyl-4,5,6,7-tetrahydro-1H-[1,2,3]triazolo[4,5-c]pyridines Enables the Discovery Synthesis and Preclinical Profiling of a P2X7 Antagonist Clinical Candidate. J Med Chem. 2018;61(1):207-23. doi:10.1021/acs.jmedchem.7b01279.
- Recourt K, van der Aart J, Jacobs G, de Kam M, Drevets W, van Nueten L, et al. Characterisation of the pharmacodynamic effects of the P2X7 receptor antagonist JNJ-54175446 using an oral dexamphetamine challenge model in healthy males in a randomised, double-blind, placebo-controlled, multiple ascending dose trial. J Psychopharmacol (Oxf). 2020;34(9):1030-42. doi:10.1177/0269881120914206.
- Rat P, Olivier E, Tanter C, Wakx A, Dutot M. A fast and reproducible cell- and 96-well plate-based method for the evaluation of P2X7 receptor activation using YO-PRO-1 fluorescent dye. J Biol Methods. 2017;4(1):e64. doi:10.14440/jbm.2017.136.
- Calzaferri F, Narros-Fernández P, de Pascual R, de Diego AMG, Nicke A, Egea J, et al. Synthesis and Pharmacological Evaluation of Novel Non-nucleotide Purine Derivatives as P2X7 Antagonists for the Treatment of Neuroinflammation. J Med Chem. 2021;64(4):2272-90. doi:10.1021/acs.jmedchem.0c02145.
- Beamer E, Morgan J, Alves M, Menéndez Méndez A, Morris G, Zimmer B, et al. Increased expression of the ATP-gated P2X7 receptor reduces responsiveness to anti-convulsants during status epilepticus in mice. Br J Pharmacol.2022;179(12):2986-3006. doi:10.1111/bph.15785.
- Mut-Arbona P, Huang L, Baranyi M, Tod P, Iring A, Calzaferri F, et al. Dual Role of the P2X7 Receptor in Dendritic Outgrowth during Physiological and Pathological Brain Development. J Neurosci Off J Soc Neurosci. 2023;43(7):1125-42. doi:10.1523/JNEUROSCI.0805-22.2022.
- Rodríguez-Ramírez KT, Norte-Muñoz M, Lucas-Ruiz F, Gallego-Ortega A, Calzaferri F, García-Bernal D, et al. Retinal response to systemic inflammation differs between sexes and neurons. Front Immunol. 2024;15:1340013. doi:10.3389/fimmu.2024.1340013.
- Valencia I, Pastor-Martínez A, Decouty-Pérez C, Lopez-Rodriguez AB, Álvarez-Rubal M, Ramos E, et al. Pharmacological evaluation of non-nucleotide purine derivatives as P2X7 antagonists for the treatment of neuroinflammation in traumatic brain injury. Br J Pharmacol. 2025;182(19):4693-709. doi:10.1111/bph.70108.
