1. INTRODUCCIÓN
1.1. Fructosa
La fructosa es un monosacárido que se encuentra de forma natural en frutas, otros vegetales o en la miel. Sin embargo, durante las últimas décadas su uso a nivel industrial ha experimentado un aumento significativo, ya sea en forma de jarabe de maíz rico en fructosa (JMAF) o sacarosa, para edulcorar multitud de alimentos altamente procesados, refrescos y bebidas azucaradas. Por ello, la fructosa se ha convertido en una de las principales fuentes de azúcares añadidos en la dieta en países desarrollados. Además, es importante destacar que la ingesta diaria de azúcares añadidos en EE.UU. supera la cantidad recomendada por la Organización Mundial de la Salud (OMS) (2018) (2).
El JMAF se desarrolló y empezó a producirse de forma industrial en la década de 1960. En el mercado existen distintos tipos de JMAF, siendo los más utilizados el HFCS-42 y el HFCS-55, que contienen un 42% y 55% de fructosa, respectivamente (3). Estos JMAF se introdujeron en sustitución de otros edulcorantes, principalmente la sacarosa, que es un disacárido formado por una fructosa y una glucosa utilizado como endulzante tradicional. Las ventajas del JMAF frente a la sacarosa son su elevada solubilidad en alimentos procesados y un menor coste de producción (4).
Todos estos factores han promovido un aumento exponencial en el consumo de fructosa. Según el Departamento de Agricultura de EEUU, el consumo de JMAF aumentó >1000% entre 1970 y 1990 y en 2004 representaba >40% de los edulcorantes calóricos en refrescos en EEUU. Aunque esta tendencia creciente hoy en día se está atenuando (5).
1.2. Enfermedades relacionadas con el consumo de fructosa
En las últimas décadas, el estilo de vida, el nivel socioeconómico y los hábitos higiénico-dietéticos han experimentado cambios en la población mundial que han promovido la adquisición de hábitos poco saludables, un estilo de vida sedentario y una occidentalización de la dieta. La dieta occidental o Western diet se caracteriza por una ingesta alta de sal, azúcares añadidos y ácidos grasos saturados (6), además de un bajo consumo de verduras y frutas frescas (7,8).
Utilizando como valor de referencia el índice de masa corporal (IMC), la OMS determinó en 2016 que las tasas de sobrepeso y obesidad se habían triplicado desde 1975, estimando que el 39% de la población mundial adulta padece sobrepeso y el 13% obesidad. Estos datos lógicamente varían entre países, siendo EE.UU. el que encabeza lalista (9).
En múltiples estudios epidemiológicos se ha demostrado que el aumento significativo del consumo de fructosa en forma JMAF como edulcorante es un factor etiológico y está relacionado con el aumento de la obesidad y alteraciones metabólicas asociadas a una respuesta inflamatoria y estrés oxidativo (10) o deterioro cardiaco (11). Además, es un factor de riesgo potencial para el desarrollo de enfermedades como el síndrome metabólico.
El síndrome metabólico (MetS) o síndrome X es un conjunto de cinco factores clínicos y metabólicos de riesgo para la salud como son la resistencia a la insulina o hiperglucemia, dislipemia (hipertrigliceridemia), hipertensión arterial, obesidad abdominal y disminución de colesterol en las HDL. Para el diagnóstico de MetS se requiere la presencia de al menos tres de las cinco alteraciones anteriormente descritas. La presencia de síndrome metabólico a su vez incrementa significativamente el riesgo de enfermedades a largo plazo como la diabetes tipo II o enfermedades cardiovasculares (12, 13).
Por otra parte, el consumo de fructosa también se ha relacionado con el desarrollo de la enfermedad del hígado graso no alcohólico (NAFLD) ya que estimula la lipogénesis de novo y aumenta el contenido de grasa hepática. Esta NAFLD se caracteriza por la acumulación de triglicéridos hepáticos, puede conducir a fibrosis hepática, cirrosis o carcinoma hepatocelular y, además, también está asociada al síndrome metabólico (8,9).
1.3. Tagatosa
La tagatosa es una cetohexosa, epímero de la fructosa, que tiene un aporte calórico menor a esta y que por ello, se utiliza como edulcorante bajo en calorías en bebidas, yogures y dulces dietéticos, entre otros (11). Tras la realización de estudios de toxicidad y seguridad en humanos y otros animales, la tagatosa fue declarada como una sustancia “Generalmente Reconocida Como Segura (GRAS)” por parte de la FDA, lo que permitió su uso a nivel industrial (14,15).
El poder edulcorante de la tagatosa equivale a un 92% del de la sacarosa (16,17). Sin embargo, posee un índice glucémico bajo y tiene un efecto hipoglucemiante si es administrada antes de una ingesta oral de glucosa (14,18,19). Además, estudios previos sugieren claramente que el consumo crónico de tagatosa no induce las mismas alteraciones metabólicas nocivas que las provocadas por el consumo crónico de fructosa, como las relacionadas con el estrés oxidativo y el efecto proinflamatorio selectivo (20). También se ha demostrado que la exposición crónica a la tagatosa produce una menor susceptibilidad del miocardio a lesión por I/R (Isquemia/Reperfusión) en comparación con la fructosa (11).
1.4. Estructura y metabolismo de la fructosa, glucosa y tagatosa
La glucosa, la fructosa y la tagatosa comparten el mismo peso molecular y fórmula química. Sin embargo, su conformación estructural es distinta (Figura 1). La fructosa es un isómero estructural de la glucosa y difieren en su grupo carbonilo. Mientras que la fructosa presenta un grupo cetona en el Carbono número 2, la glucosa presenta un grupo aldehído en el carbono número 1. Por otro lado, la tagatosa es un epímero de la fructosa que cambia su configuración espacial en el cuarto carbono, que es quiral.
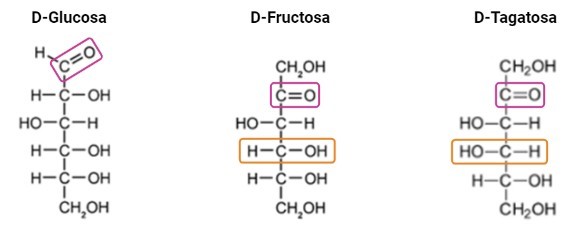
Figura 1. Estructura de la D-glucosa, D-fructosa y D-tagatosa. En morado se señalan la diferencia entre el grupo aldehído de la glucosa y el grupo ceto de la fructosa y la tagatosa. En naranja se señala la diferencia en la conformación espacial en el C4 quiral entre la fructosa y la tagatosa.
Pese a que los tres monosacáridos presentan una conformación similar (Figura 1), sus diferencias estructurales condicionan distintas vías de absorción a través del intestino y, además, distintas vías de metabolizarse en el organismo (Figura 2).
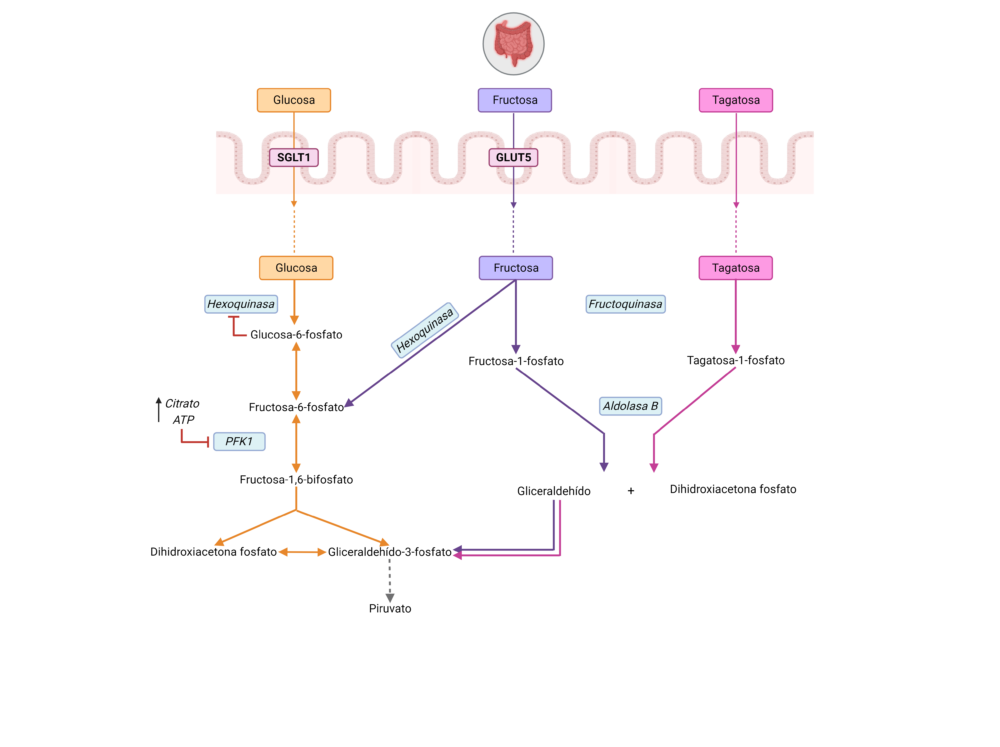
Figura 2. Ruta metabólica y absorción de la D-glucosa, D-fructosa y D-tagatosa en el organismo. La vía de color naranja corresponde a la vía metabólica de la glucosa, la vía de color morado al de la fructosa y la vía de color rosa al de la tagatosa. Las enzimas están señaladas en un recuadro azul en cursiva y los transportadores de glucosa y fructosa en un recuadro rojo. Figura creada con BioRender.com
En primer lugar, la glucosa se absorbe a nivel intestinal en los enterocitos a través de SGLT1, que es un cotransportador sodio-glucosa localizado en la membrana apical y que es dependiente de un gradiente de sodio generado por una Na+-K+ ATPasa. Una vez pasa a circulación mediante GLUT2 se distribuye a distintos tejidos, donde se va a llevar a cabo la glucólisis para metabolizar la glucosa a dos moléculas de piruvato y generar ATP. La primera reacción de esta ruta está catalizada por una hexoquinasa que fosforila la glucosa en la posición 6 utilizando ATP. Esta reacción está muy desplazada hacia la formación de G6P y se considera irreversible, esto hace que sea un punto de regulación de la ruta, aunque no el principal.
La segunda reacción la lleva a cabo la glucosa fosfato isomerasa, transforma a la glucosa 6-P en fructosa-6-P. A continuación, este azúcar vuelve a fosforilarse en el primer carbono para dar lugar a la fructosa-1,6-bifosfato gracias a la acción de la enzima PFK1 (fosfofructoquinasa 1), cuya reacción es irreversible y es el punto principal de regulación de la glucólisis ya que se inhibe por ATP (producto de la vía glucolítica) y por citrato (intermediario del ciclo del ácido cítrico, CAC). A partir de aquí, la fructosa-1,6-bifosfato se escinde en dihidroxiacetona fosfato (DHP) y gliceraldehído-3-fosfato (GAP) por una aldolasa. Pese a que la reacción no está favorecida energéticamente, el hecho de que el GAP se use inmediatamente tras la escisión hace que la reacción esté favorecida hacia la ruptura de la fructosa-1,6-bifosfato. El gliceraldehido-3-fosfato posteriormente se convertirá en piruvato tras una serie de reacciones (21, 22) (Figura 2).
Por otro lado, la fructosa se absorbe en el intestino a través del transportador GLUT 5, el único transportador específico para la fructosa sin capacidad para transportar glucosa o galactosa (23, 24). Accede a la circulación sistémica gracias a través de la membrana basolateral gracias a GLUT2, al igual que la glucosa. Cuando la fructosa llega a los distintos tejidos se metaboliza de forma distinta a la glucosa, mediante la fructólisis, que se limita principalmente al hígado, no como la glucólisis que se lleva a cabo en todas las células del organismo, aunque comparte enzimas e intermediarios metabólicos con esta.
La primera reacción que experimenta la fructosa en su metabolismo a través de la fructólisis es la fosforilación a fructosa-1-fosfato a través de una cetohexoquinasa (KHK), conocida también como fructoquinasa en el hígado. Esta fructosa-1-fosfato a través de la aldolasa B se escinde en dihidroxiacetona fosfato (DHAP) y gliceraldehido (GA). Tanto la DHAP como el GA se pueden metabolizar mediante una vía compartida con la glucólisis a gliceraldehido-3-fosfato (GA3P). Por otra parte, el GA se puede reducir y fosforilar para dar lugar al glicerol-3-fosfato (G3P), que sirve de sustrato para la síntesis de lípidos o transporte de equivalentes reductores a las mitocondrias a través de la lanzadera de G3P (25, 26).
Además, la fructosa en los tejidos extrahepáticos puede introducirse en la glucólisis fosforilándose a F6P a través de una hexoquinasa, saltándose el primer punto de regulación de la vía glucolítica. No obstante, en su vía metabólica mayoritaria a través de la cetohexoquinasa (KHK) o fructoquinasa se salta además el punto principal de regulación de la glucólisis, catalizado por la PFK1, enzima limitante de velocidad de la vía glucolítica y regulada, tanto por altos niveles de ATP como de citrato (26). La KHK es una enzima presente sobre todo en intestino e hígado y, por tanto, la fructólisis es una vía típica en esos tejidos, mientras que la otra ruta alternativa del metabolismo de la fructosa es más habitual en el resto de los tejidos extrahepáticos (Figura 2).
Por último, de la tagatosa ingerida por vía oral se absorbe aproximadamente un 20% y se metaboliza principalmente a través del hígado (17, 19). La tagatosa a la hora de metabolizarse sigue la misma vía que la fructosa (27), se transforma a tagatosa-1-fosfato (T1P) a través de la fructoquinasa, y, del mismo modo, la aldolasa B hidroliza la tagatosa-1-fosfato a gliceraldehido y dihidroxiacetona fosfato (Figura 2).
Al igual que la fructosa-1-fosfato, el aumento de tagatosa-1-fosfato actúa como activador de la glucoquinasa (la hexoquinasa de la glucólisis hepática), lo que conlleva una mayor transformación de glucosa a glucosa-6-fosfato y esto activa aún más a la glucógeno sintasa. Así mismo, hay estudios que sugieren que la tagatosa-1-fosfato, de forma similar a la fructosa-1-fosfato, inhibe a la glucógeno fosforilasa. De tal forma que la presencia tanto de fructosa-1-fosfato como de tagatosa-1-fosfato aumenta la síntesis de glucógeno y disminuye su utilización (27).
1.5. Hormonas tiroideas
Las hormonas tiroideas son esenciales para el correcto desarrollo, crecimiento, diferenciación celular o regulación metabólica del organismo (28). La glándula tiroides es la encargada de la producción de las hormonas tiroideas. Esta glándula consta de folículos en los que la hormona se sintetiza a través de la adición de yodo a residuos de tirosina de la tiroglobulina (29). Así se producen y liberan tanto triidotironina (T3) como tiroxina (T4), todo ello regulado por la hormona de la hipófisis estimuladora del tiroides o TSH. La mayoría de la T4 producida por la glándula tiroides circula en sangre unida a una proteína transportadora (TGB, del inglés Thyroxine-binding globulin), y solo una pequeña fracción circula libre. Esta pequeña proporción de hormona libre regula mediante un mecanismo de feedback negativo la secreción de TSH (30, 31).
La acción molecular y unión a los receptores nucleares de hormonas tiroideas (THR) está íntimamente relacionada con la cantidad de hormona activa disponible, tanto a nivel plasmático como ya luego a nivel tisular. El transporte de la T4 libre circulante al interior celular se produce principalmente a través de varios receptores de la familia del transportador de monocarboxilatos, como MCTP8 o MCTP10 (32), receptores OATP (transportador de aniones orgánicos) o LAT (transportadores de aminoácidos de tipo L) (33). Una vez en el interior, la T4 se transforma en la forma activa de la hormona tiroidea, la T3, a través de las enzimas desyodasas (en inglés deiodinases) (DIO) (34, 35). De estas enzimas hay varios tipos, siendo la DIO1 y, sobre todo, la DIO2 las encargadas de la activación de las hormonas tiroideas, mientras que la DIO3 parece que tiene más bien un papel de degradación y inactivación de dicha hormona (28).
Entre las múltiples acciones de la hormona tiroidea activa (T3) se encuentra su participación en la regulación de la transcripción de múltiples genes. La T3 se une a su receptor específico THR (que puede ser de tipo alfa o beta), el cual queda activado para ejercer su papel de factor de transcripción, uniéndose al promotor de sus genes diana (los que tengan la secuencia TRE, elemento respuesta a hormonas tiroideas) y regulando así su expresión (Figura 3). De entre esos genes diana y en los que se ha centrado este estudio, se encuentran las proteínas desacoplantes o UCP2 y UCP3 (del inglés Uncoupling Protein). Las proteínas desacoplantes o termogeninas están localizadas en la membrana mitocondrial interna y actúan como mecanismo de regulación mediante el cual la energía se utiliza para generar calor y/o evitar la saturación de la cadena respiratoria. En concreto, la mayoría de los autores proponen que UCP2 y UCP3 actúan regulando la cantidad de ATP disponible en las células, el flujo de electrones en la cadena respiratoria, la producción de ROS mitocondriales, así como también protegen contra el daño celular y disminuyen la secreción de insulina (36, 37). Todo el proceso descrito anteriormente desde la hormona inactiva T4 libre en plasma hasta la expresión de UCP se describe en la Figura 3.
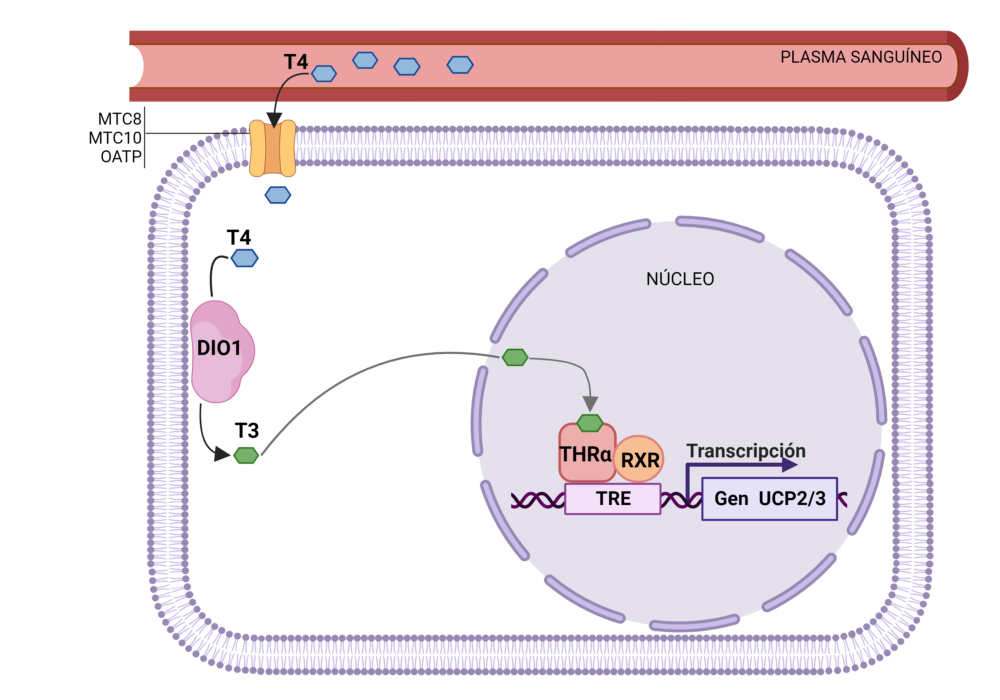
Figura 3. Proceso de activación de la transcripción de UCP2/3 a partir de la T4 libre en plasma. La T4 libre se introduce en la célula a través de transportadores (MTC8, MTC10 y OATP), se activa por DIO1 a T3. La T3 accede al núcleo celular, se une al receptor (THRa) que forma un dímero con el receptor RXR (receptor X retinoide), los cuales se unen a la región TRE del gen diana y activan su transcripción.
Diversos estudios han demostrado que la nutrición puede influir en la fisiología del tiroides. Así, por ejemplo, una dieta rica en grasa (HFD, del inglés High Fat Diet) provoca cambios en el perfil lipídico, el funcionamiento del tiroides e incluso en su morfología. Es más, dietas con un elevado contenido en carbohidratos también parecen afectar al funcionamiento de las hormonas tiroideas y a sus niveles en sangre (38).
1.6. Programación fetal
La hipótesis acerca de la programación fetal la propusieron Barker y Hales en 1992 (39), donde se postula que la susceptibilidad a las enfermedades crónicas de un adulto se presenta en respuesta a un entorno adverso nutricional prenatal y/o perinatal. Esta hipótesis ha sido apoyada por múltiples estudios epidemiológicos (40).
La programación fetal es un proceso mediante el cual diversos factores ambientales, genéticos y el ambiente nutricional intrauterino recibido durante el desarrollo embrionario y fetal van a determinar el desarrollo posterior de enfermedades en la edad adulta. Estos factores pueden dar lugar a alteraciones estructurales, fisiológicas y metabólicas permanentes durante el desarrollo y en la edad adulta de la descendencia (41). Hay estudios que afirman y constatan que el consumo de fructosa durante el embarazo produce un fenotipo programado que conlleva efectos perjudiciales en la descendencia (42).
Esta exposición a factores ambientales y del entorno materno puede provocar, a través de la programación fetal, cambios epigenéticos que conducirán a alteraciones en la expresión de algunos genes. Es más, es importante resaltar que la programación fetal permanece activa generación tras generación cuando la exposición al factor de estrés ocurre durante períodos críticos de la vida, como el embarazo y la lactancia (43).
2. OBJETIVOS
Con los antecedentes previamente descritos, quisimos averiguar si había un componente nutrigenómico que afectara a la funcionalidad y metabolismo de las hormonas tiroideas, para lo cual evaluamos el efecto de diversas dietas: fructosa, tagatosa y Western diet. Además, con el fin de estudiar la posible implicación de la programación fetal, nos planteamos determinar si los efectos observados eran dependientes de la dieta materna.
Para ello, nos planteamos los siguientes objetivos:
- Estudiar si la ingesta de fructosa durante 21 días en ratas tiene algún efecto sobre la concentración de T4 libre en el plasma y, en consecuencia, sobre la expresión génica de la desyodasa y los genes diana de la hormona tiroidea: UCP2 y UCP3 en hígado, tejido adiposo lumbar e íleon.
- Evaluar si la ingesta de tagatosa genera los mismos efectos que la fructosa en la ruta descrita en el apartado anterior, para comprobar si los efectos observados son específicos de alguno de estos 2 azúcares.
- Determinar si la ingesta de una dieta Western (u occidental) (rica en fructosa y colesterol) induce los mismos efectos que la ingesta de fructosa sola en la ruta mencionada en el primer objetivo, para comprobar si la presencia de colesterol potencia los efectos provocados por la fructosa.
- Valorar si los efectos observados están influenciados por la dieta materna y, por lo tanto, implican la intervención de procesos de programación fetal. Para ello, a las madres de los animales utilizados en los tres anteriores apartados se les suministró en la gestación agua sin aditivos o agua suplementada con fructosa.
3. MATERIAL Y MÉTODOS
3.1. Diseño experimental
El diseño experimental que se llevó a cabo se detalla a continuación (Figura 4). Ratas Sprague-Dawley se aparearon y una vez se constató el inicio de la gestación, las hembras (generación F0) recibieron una dieta estándar y se separaron en dos grupos según la bebida administrada durante este periodo. Uno de los grupos, el grupo de madres-Control, recibió agua sin ningún aditivo, mientras que el otro grupo de ratas gestantes, grupo de madres-Fructosa, recibió agua con una solución de fructosa al 10% p/v.
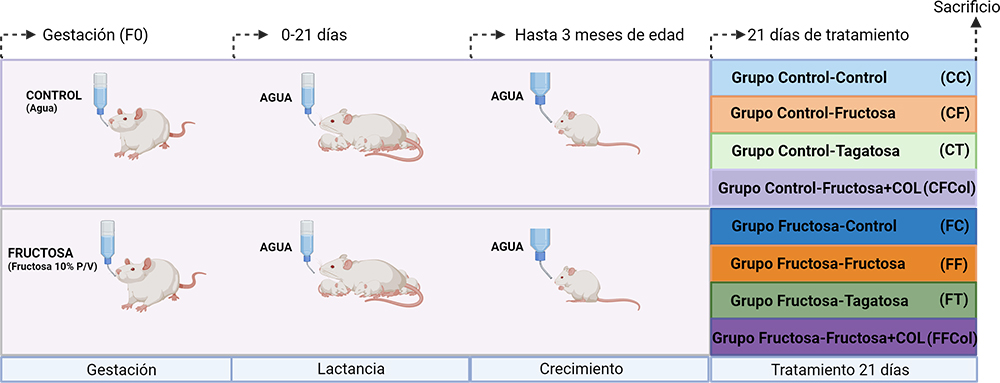
Figura 4. Diseño experimental. Creada con BioRender.com
Tras el parto, tanto la madre como los descendientes recibieron en la bebida agua sin ningún aditivo en ambos grupos hasta el inicio de la segunda parte del estudio (a los 3 meses de edad). Solo se prosiguió el estudio con los descendientes macho (generación F1). A los 3 meses de edad todos los descendientes, procedentes tanto de madres-control como de madres-fructosa, se separaron en cuatro grupos en función de la dieta recibida:
- Grupo Control: recibieron una dieta sólida estándar y agua sin ningún aditivo.
- Grupo de Fructosa: recibieron una dieta sólida estándar y agua con una solución de fructosa al 10% p/v.
- Grupo de Tagatosa: recibieron una dieta sólida estándar y agua con una solución de tagatosa al 10% p/v.
- Grupo de fructosa y colesterol: recibieron una dieta sólida estándar suplementada con un 2% de colesterol y agua suplementada con fructosa al 10% p/v (Figura 4).
Así, se obtuvieron un total de 8 grupos experimentales (Figura 4). La primera letra de cada grupo indica el tratamiento recibido por las madres y la segunda letra el tratamiento recibido por los descendientes. Tras 21 días de tratamiento las ratas fueron sacrificadas. La sangre se recogió en tubos Vacutainer® con EDTA-K2. Para la obtención del plasma se centrifugaron las muestras de sangre (20 min, 3000rpm, 4ºC), se repartieron en diferentes alícuotas y se almacenaron a -80ºC hasta el momento de la medición de los parámetros. Los tejidos obtenidos fueron congelados en nitrógeno líquido tras su obtención y se almacenaron a -80ºC hasta el momento de la medición de los parámetros correspondientes.
3.2. Ensayos de parámetros plasmáticos
3.2.a. Determinación de T4 libre en plasma
La determinación cuantitativa de la T4 libre en plasma se hizo con un kit de NovaTech (Immundiagnostica GmbH), que permite la detección mediante un método inmunoenzimático y colorimétrico de T4 libre, tanto en suero como en plasma. Este protocolo se basa en la realización de un ELISA, que en este caso era tipo sándwich.
Para ello se pipetearon en cada pocillo 50mL de cada muestra, 6 soluciones estándar a diferentes concentraciones (A: 0.0 ng/l; B: 3.0 ng/l; C:9.5 ng/l; D: 21.0 ng/l; E: 36.0 ng/l; F: 70 ng/l) y de control, a los que se añadieron posteriormente 100mL de reactivo conjugado (T4-HRP). A continuación, se incubó la mezcla de reacción 1h a 37ºC en oscuridad. Tras el tiempo de incubación se lavó cada pocillo tres veces con 300 mL de una solución de lavado. Seguidamente, se añadieron 100mL de solución sustrato (TMB) y se incubó en oscuridad a 20-25ºC durante 15 minutos, tras los cuales se añadió una solución de parada. Por último, se midió la absorbancia a 450 nm en un lector de placas SpectroStar Nano (BMG Labtech, Alemania).
Con la absorbancia obtenida de los estándares se realizó una recta de calibrado a partir de la cual se interpolaron las absorbancias de las muestras y el control.
3.3. Ensayos de parámetros moleculares
3.3.a. Homogeneización de los tejidos y extracción de ARN
Para la homogenización de los tejidos se pesaron 100mg de muestra a los que se les añadió Tri Reagent® (Invitrogen, ThermoFisher, EEUU) y se llevó a cabo la homogeneización mediante la utilización de un TissueLyser LT (Qiagen, EEUU). Se centrifugó posteriormente el homogenado (10 min, 16000g, 4Cº). A continuación, para aislar el ARN se utilizó bromocloropropano y se volvieron a centrifugar las muestras (10 min, 16000g, 4Cº) pudiendo así extraer el ARN que quedó disuelto en la fase acuosa.
3.3.b. Purificación del ARN. Cuantificación, determinación del grado de pureza e integridad del ARN. Tratamiento con DNasa
Tras la extracción, el ARN se purificó mediante el kit RibopureTM (Invitrogen, ThermoFisher, EEUU). Para llevar a cabo la cuantificación del ARN obtenido se utilizó un NanoDrop® One (ThermoFisher, EEUU). Además, se determinó el grado de pureza del ARN extraído por duplicado teniendo en cuenta los siguientes parámetros:
- Relación de absorbancias a 260 y 230nm para detectar una posible contaminación de la muestra con solventes orgánicos.
- Relación de absorbancias a 260 y 280nm para evaluar una posible contaminación con proteínas.
El grado de pureza de las muestras se consideró óptimo cuando la relación de absorbancias se encontraba en el intervalo 1.8-2.0. La integridad del ARN obtenido se determinó mediante una electroforesis (90V, 20 min) en gel de agarosa al 2% teñido con Midori Green Advance (Nippon Genetics) y para ello se cargaron 500 ng de ARN con tampón de carga y agua, en los que se observaron las bandas correspondientes al ARN ribosomal (18S y 28S), tal y como se observa en la (Figura 5).
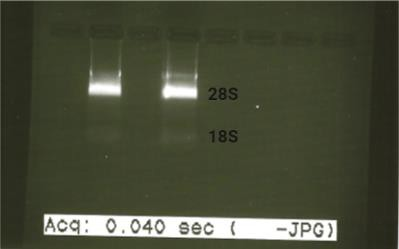
Figura 5. Comprobación de la integridad del ARN obtenido mediante electroforesis en gel de agarosa. Bandas correspondientes al ARN ribosomal 18S y 28S.
Para retirar las posibles trazas de ADN contaminante y evitar así que interfiera en los posteriores análisis, se llevó a cabo un tratamiento con DNasa, mediante el kit Turbo DNA-freeTM (Invitrogen, ThermoFisher, EE.UU.) y se determinó de nuevo la concentración de ARN tras el tratamiento, siguiendo el protocolo mencionado anteriormente. Se realizó posteriormente una PCR (reacción en cadena de la polimerasa) para comprobar la ausencia de ADN en las muestras utilizando los cebadores de un gen constitutivo: el RPS29.
3.3.c. Retrotranscripción
Para realizar los ensayos de expresión génica requeridos en este estudio fue necesario obtener el ADN complementario (ADNc) a partir del ARNm, para lo cual se llevó a cabo una retrotranscripción utilizando el kit SuperScript® II Reverse Transcriptase (Invitrogen, EEUU) y un Termociclador iCycler (BioRad, EEUU). Para ello se utilizaron 2,5 ng de ARN tratado con DNasa en muestras de hígado e íleon y 1 ng de ARN en muestras de tejido adiposo. Se comprobó la funcionalidad del ADNc obtenido mediante una PCR (reacción en cadena de la polimerasa) utilizando cebadores del gen RPS29 como referencia y una electroforesis posterior (Figura 6).
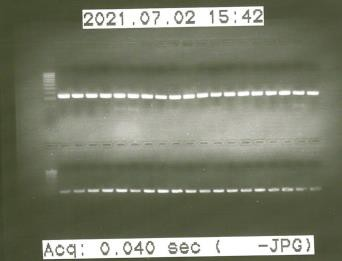
Figura 6. Comprobación de la funcionalidad del cDNA mediante electroforesis en gel de agarosa. Bandas correspondientes al producto de amplificación de los cebadores correspondientes a RPS29.
3.3.d. Estudio de la expresión génica
Se utilizó la técnica de PCR a tiempo real o Q-PCR para el estudio de la expresión génica. La PCR a tiempo real permite tanto el análisis cualitativo como cuantitativo simultáneo de las muestras. Para ello emplea la medida de la fluorescencia en cada uno de los ciclos de amplificación ya que el ADN amplificado en cada ciclo queda marcado con SyBR- Green I, un fluoróforo específico de ADN bicatenario, siendo la fluorescencia proporcional a la cantidad de producto amplificado (44).
3.3.1.a. Diseño de primers
Los cebadores (primers) se diseñaron utilizando el programa Primer Blast estableciendo los siguientes parámetros: un tamaño de 100 a 300 pb de producto de amplificación y una Tm de 60ºC. La secuencia de los cebadores se comprobó mediante el programa Beacon Designer. Se verificó que los cebadores eran válidos buscando si amplificaban de forma específica, tanto el primer Forward como el primer Reverse, en la secuencia FASTA del gen correspondiente. La secuencia de los cebadores utilizados se muestra en la tabla 1.
Tabla 1. Secuencia de los primers (cebadores) F y R de los genes DIO1, THRA1, UCP3 y UCP2 con su procedencia.

3.3.2.a. Recta de calibrado
El estudio de la expresión génica y cuantificación del ADNc requiere de una recta de calibrado en la que interpolar los valores obtenidos posteriormente en los ensayos de las muestras. Para ello, a partir de concentraciones conocidas del ADNc diana, se realizaron diluciones seriadas 1/10 (50, 5, 0.5 y 0.05 ng/μL). La eficiencia de los primers (E) se calculó a partir de los Cp de la recta de calibrado obtenida y se realizó una electroforesis en gel de agarosa para comprobar que el tamaño del producto de amplificación era el esperado y no se observaban productos secundarios (Figura 7).
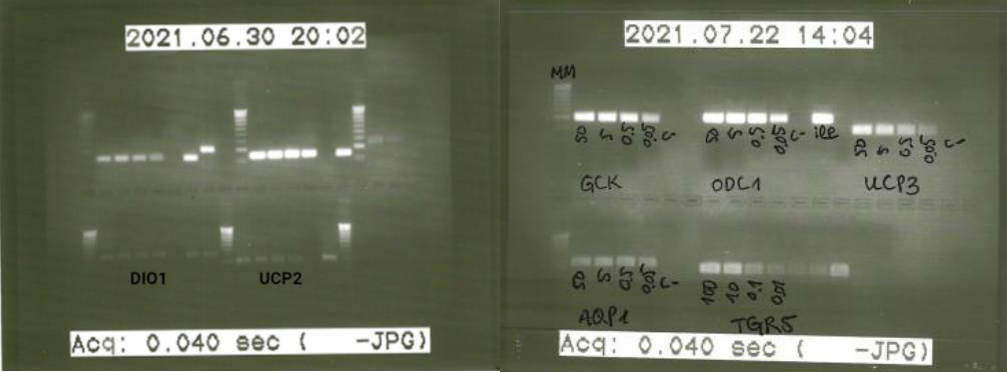
Figura 7. Electroforesis en gel de agarosa de las rectas de calibrado de DIO1, UCP2 y UCP3.
Además, se pudo verificar la amplificación de la secuencia diana mediante la determinación de su Tm (temperatura de fusión) ya que este parámetro depende de la longitud del producto de amplificación y el porcentaje de GC.
3.3.3.a Protocolo PCR a tiempo real para la cuantificación de la expresión génica
La PCR a tiempo real se llevó a cabo en un Termociclador CFX96 Touch Real-Time PCR Detection System (BioRad, EEUU) con los cebadores descritos anteriormente. Para ello se utilizó una dilución 1/30 de ADNc en muestras de hígado e íleon y una dilución 1/5 en muestras de tejido adiposo lumbar (TAL) alcanzando así una concentración próxima a 5ng/μL con el fin de asegurar que los valores de Cp se encontrasen dentro de la recta de calibrado y no superasen valores superiores a 33 ciclos, a partir de los cuales hay probabilidad de que amplifiquen productos inespecíficos y se pierde eficiencia.
Para cada muestra, los volúmenes de reactivos añadidos fueron: 6mL de agua mili-Q estéril, 1μL de cebador F (10μM), 1μL de cebador R (10μM), 2μL de muestra de ADNc y 10μL de SuBR Green Premix Ex Taq (Takara Bio Inc., Japón). La cuantificación de la expresión génica se basa en el método 2-DCt o método de Pfaffl (45). Para el cálculo se utilizó RPS29 como gen de referencia, cuya expresión no varía en función de las condiciones experimentales. En el caso del tejido adiposo el gen de referencia utilizado fue el HPRT1.
3.4. Análisis estadístico
En el análisis estadístico los resultados se expresaron como media ± error estándar (S.E.) y corresponden a 7-8 animales por grupo. Los efectos del tratamiento fueron analizados mediante análisis de varianza de doble vía (ANOVA). Cuando las diferencias fueron estadísticamente significativas (p<0,05) se analizaron mediante la corrección de Bonferroni para identificar la significancia de cada una de las varianzas. Toda la determinación estadística se realizó utilizando el programa informático SPSS (versión 27).
Así, los resultados estadísticamente significativos se indican:
- Con almohadillas (#) si se presentan diferencias estadísticamente significativas (# P<0.05; ## P<0.01; ### P<0.001) entre los descendientes tratados con la misma dieta durante su edad adulta (21 días), pero procedentes de madres tratadas con diferente dieta durante la gestación (Factor madre: M; Madres control vs. Madres fructosa).
- Con asteriscos (*) si se presentan diferencias estadísticamente significativas (* P<0.05; ** P<0.01; *** P<0.001) entre los descendientes tratados con diferente dieta durante su edad adulta (21 días), pero procedentes de madres tratadas con la misma dieta durante la gestación (Factor dieta: D).
- El valor de η2 determina la proporción de varianza atribuida a la influencia de la madre (M), de la dieta en la descendencia (D) o a la interacción entre ambos parámetros (M*D).
4. RESULTADOS Y DISCUSIÓN
Para facilitar la organización y el análisis de los resultados se han dividido en dos estudios según la dieta de la descendencia en la edad adulta (fructosa versus tagatosa; fructosa versus Western diet) y el tejido implicado (hígado, íleon y TAL).
4.1. Efectos de la fructosa vs tagatosa
4.1.a. T4 libre en plasma
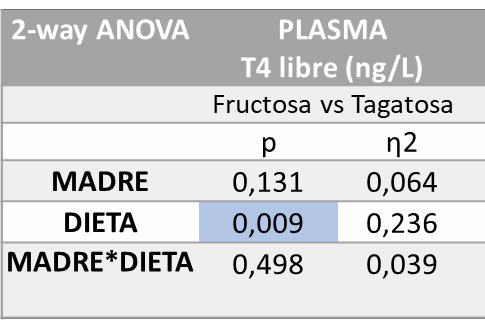
La Tabla 2 muestra los resultados del análisis estadístico de varianza de doble vía (ANOVA) en cuanto a la concentración de T4 libre en plasma. Se puede observar que hay diferencias estadísticamente significativas (p=0,009) en función de la dieta recibida por la descendencia (machos F1).
Tabla 2. ANOVA de doble vía T4 libre en plasma fructosa vs tagatosa. P valor: indica la probabilidad de obtener un valor tan o más extremo al observado, partiendo de la suposición de la igualdad de efecto que marca la hipótesis nula. η2: determina la proporción de varianza atribuida a la influencia de la madre (M), a de la dieta en la descendencia (D) o a la interacción entre ambos parámetros (M*D).
Tras la corrección a posteriori de Bonferroni, se muestran los datos obtenidos de la T4 libre en plasma en la Figura 8. Se observa que hay diferencias estadísticamente significativas de los niveles de T4 libre en plasma entre los descendientes macho que tomaron fructosa en el agua de bebida (FF) frente a los que recibieron tagatosa (FT), siendo la dieta de la madre la misma (Madres-Fructosa). Así, el grupo de descendientes que ingirió tagatosa (FT) mostró un aumento significativo frente al suplementado con fructosa, pero sin alcanzar valores significativos frente a los que tomaron agua sin aditivos (FC). Curiosamente, en los descendientes de madres control no se observaron diferencias significativas entre los tres grupos dietéticos. Estos resultados indican que la ingesta de tagatosa y no de fructosa aumenta la concentración de hormona T4 libre en plasma y que, además, es un efecto dependiente de la dieta materna.
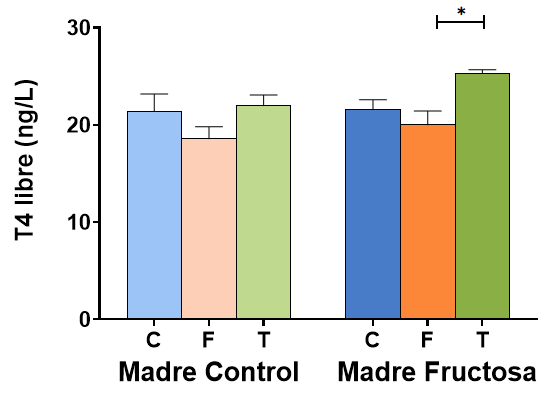
Figura 8. Concentración (ng/L) de T4 libre en plasma de machos F1 tras los 21 días de tratamiento (barra azul: agua-C; barra naranja: fructosa-F; barra verde: tagatosa-T). Las barras más claras pertenecen a los machos F1 descendientes de madres control (recibieron agua sin aditivos durante la gestación), mientras que las oscuras pertenecen a machos F1 descendientes de madres fructosa (recibieron fructosa como tratamiento en el agua de bebida durante la gestación). Los datos presentados son las medias ± error estándar (S.E.). El símbolo * indica diferencias estadísticamente significativas entre dietas, y el # entre la ingesta materna.
Estudios anteriores han descrito que la ingesta de fructosa puede disminuir la captación de T4 por el hígado, con lo que habría un incremento en plasma, y ese efecto podría estar relacionado con el aumento de norepinefrina observado tras una dieta rica en carbohidratos (46). Además, sugieren que la captación tisular de T4 puede estar regulada por las reservas de energía intracelular, afectando al metabolismo y disponibilidad de la hormona, dado que parece ser que hay dependencia de ATP por parte del hígado en el transporte de yodotironinas (47). Sin embargo, en el presente estudio no se han observado cambios de T4 libre en plasma dependientes del consumo de fructosa y, por el contrario, el aumento que se ha observado se ha producido por la ingesta de la tagatosa, lo cual no está descrito en la bibliografía y menos que sea dependiente de la dieta materna. Esta falta de efecto mediado por la ingesta de fructosa puede deberse a que los resultados descritos por Hallfrisch et al. en 1983 fueron obtenidos tras un tratamiento a corto plazo y quizás estos efectos no se produzcan a largo plazo, por lo que sería necesario realizar más trabajos en este ámbito.
4.1.b. Hígado
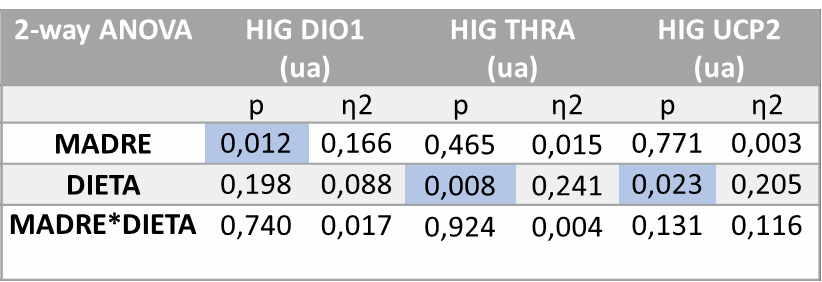
El análisis de varianza ANOVA de doble vía realizado sobre la expresión de DIO1 en el hígado (Tabla 3) muestra que se encuentran diferencias estadísticamente significativas (p=0,012) debidas a la influencia de la dieta recibida por la madre en la gestación (M). También hay diferencias estadísticamente significativas en cuanto a la expresión de THRa (p=0,008) en función de la dieta recibida por la descendencia (D). Además, se muestran diferencias estadísticamente significativas en la expresión de UCP2 (p=0.023) por la influencia de la dieta recibida por la descendencia (D).
Tabla 3. ANOVA de doble vía expresión génica de DIO1, THRa y UCP2 en hígado. P valor: indica la probabilidad de obtener un valor tan o más extremo al observado, partiendo de la suposición de la igualdad de efecto que marca la hipótesis nula. η2: determina la proporción de varianza atribuida a la influencia de la madre (M), a de la dieta en la descendencia (D) o a la interacción ent re ambos parámetros (M*D).
La expresión génica analizada en el hígado tras la corrección de Bonferroni (Figura 9) muestra un perfil muy similar en DIO1 al encontrado para los niveles plasmáticos de T4 (Figura 8), esto es, el mayor nivel de expresión se observa en los hijos de madres-fructosa que consumieron tagatosa (FT), sin alcanzar valores significativos con respecto a los descendientes que consumieron fructosa o agua sin aditivos. Sí que existen diferencias estadísticamente significativas entre ese grupo FT y los descendientes de madres control que también tomaron tagatosa (CT). Así, se observa que la tagatosa aumenta la expresión de DIO1 hepática cuando la madre consumió fructosa durante el embarazo, lo que coincide con el aumento de la T4 libre en el mismo grupo, y este efecto no se observa con el consumo de fructosa ni en los descendientes de madres control (Figura 9). Ello indicaría que en ese grupo FT no sólo está aumentada la T4 en plasma sino también su activación a T3 en el hígado y hay un efecto claro de programación fetal.
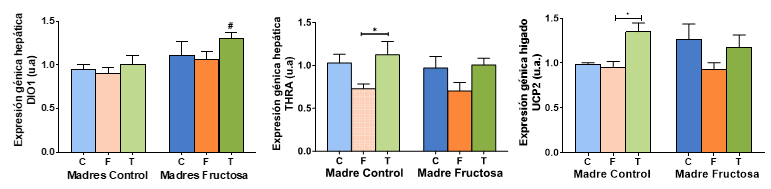
Figura 9. Expresión génica de DIO1, THRa y UCP2 en hígado de machos F1 tras los 21 días de tratamiento (barra azul: agua-C; barra naranja: fructosa-F; barra verde: tagatosa-T). Las barras más claras pertenecen a los machos F1 descendientes de madres control (recibieron agua sin aditivos durante la gestación), mientras que las oscuras pertenecen a machos F1 descendientes de madres fructosa (recibieron fructosa como tratamiento en el agua de bebida durante la gestación). Los datos presentados son las medias ± error estándar (S.E.). El símbolo * indica diferencias estadísticamente significativas entre dietas, y el # entre la ingesta materna.
En cuanto a la expresión del receptor de hormonas tiroideas (THRa), la ingesta de fructosa provocó una clara disminución, tanto en los descendientes de madres-control (CF) como en los provenientes de madres-fructosa (FF) en comparación con los restantes grupos experimentales, si bien estas diferencias sólo llegaron a ser estadísticamente significativas en los hijos de madres control suplementados con fructosa (CF) frente a los que consumieron tagatosa (CT) (Figura 9).
Curiosamente, al estudiar la expresión de UCP2, gen diana de las hormonas tiroideas, el perfil observado era más parecido al del propio receptor THRa que al de la T4 en plasma o a la expresión de la DIO1. Estos resultados refuerzan la hipótesis planteada por algunos autores que defiende que DIO1 es una enzima que activa las hormonas tiroideas, pero más con una función sistémica y circulatoria (porque trabaja a nivel de membrana) que a nivel del propio tejido donde se encuentra (que ahí sería DIO2 la más importante) (31).
Además, otros autores han descrito que la señalización mediada por hormonas tiroideas puede ocurrir sin que estas hormonas activen al receptor y, en esos casos, la modulación de los genes diana es conseguida por una mayor o menor presencia del THR correspondiente (48).
4.1.c. Íleon
Tras el análisis estadístico (ANOVA de doble vía) realizado sobre los genes (Tabla 4) se observaron diferencias estadísticamente significativas en cuanto a la expresión de UCP2 en íleon por el efecto del tratamiento de la madre recibido durante la gestación (M; p=0,021) y por el efecto de la interacción entre ambos tratamientos (M*D; p=0,043).
Tabla 4. ANOVA de doble vía expresión génica DIO1 y UCP2 íleon. P valor: indica la probabilidad de obtener un valor tan o más extremo al observado, partiendo de la suposición de la igualdad de efecto que marca la hipótesis nula. η2: determina la proporción de varianza atribuida a la influencia de la madre (M), a de la dieta en la descendencia (D) o a la interacción entre ambos parámetros (M*D).
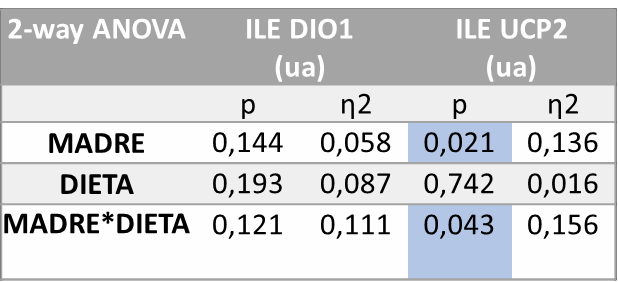
Los resultados obtenidos tras la corrección de Bonferroni se muestran en la Figura 10. En cuanto a los resultados de la expresión génica medidos en el íleon se observa que, tanto DIO1 como UCP2 presentan perfiles muy similares. Así, en DIO1 existen diferencias estadísticamente significativas entre los hijos que tomaron fructosa en el agua de bebida, siendo descendientes de madres que recibieron únicamente agua (CF) durante su gestación y los hijos de madres que recibieron fructosa en el agua de bebida (FF). Las diferencias estadísticamente significativas en la expresión de UCP2 se vuelven a observar entre los mismos grupos: CF y FF.
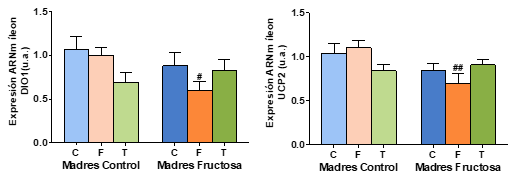
Figura 10. Expresión génica de DIO1 y UCP2 en íleon de machos F1 tras los 21 días de tratamiento (barra azul: agua-C; barra naranja: fructosa-F; barra verde: tagatosa-T). Las barras más claras pertenecen a los machos F1 descendientes de madres control (recibieron agua sin aditivos durante la gestación), mientras que las oscuras pertenecen a machos F1 descendientes de madres fructosa (recibieron fructosa como tratamiento en el agua de bebida durante la gestación). Los datos presentados son las medias ± error estándar (S.E.). El símbolo * indica diferencias estadísticamente significativas entre dietas, y el # entre la ingesta materna.
En el íleon se observa que, mientras que la tagatosa no produce cambios significativos en la expresión génica, la fructosa disminuye la expresión tanto de DIO1 y UCP2 en los descendientes de madre fructosa, pero no así en los descendientes de madre control.
Por tanto, la expresión génica en íleon sí que se ve afectada por la fructosa. En este caso se observa que los descendientes de madre fructosa que tomaron fructosa durante 21 días presentan una disminución de la expresión de DIO1, que va relacionada con la consecuente disminución de la expresión de UCP2 y no con lo observado en la T4 plasmática (Figura 8).
4.1.d. Tejido adiposo lumbar (TAL)
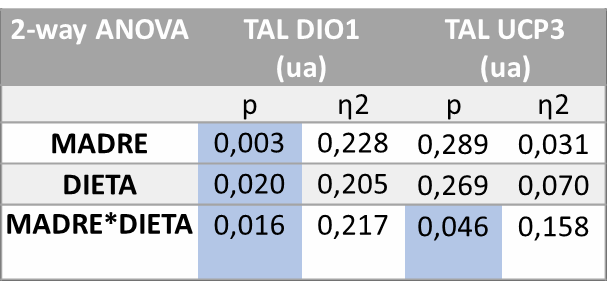
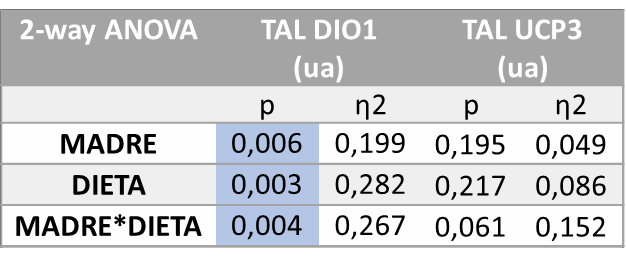
Por último, se estudió la expresión génica en el tejido adiposo lumbar. El tratamiento de los resultados mediante el análisis de varianza ANOVA de doble vía (Tabla 5) mostró que existen diferencias estadísticamente significativas en la expresión de DIO1 tanto por la influencia del tratamiento de la madre (M, p=0,003), la influencia del tratamiento recibido por la descendencia (D, p=0,020), como por la interacción entre ambos tratamientos (MD, p=0,016). También se observan diferencias estadísticamente significativas en la expresión de UCP3 debidas a la interacción entre ambos tratamientos (M*D, p=0,046).
Tabla 5. ANOVA de doble vía expresión génica DIO1 y UCP3 en TAL. P valor: indica la probabilidad de obtener un valor tan o más extremo al observado, partiendo de la suposición de la igualdad de efecto que marca la hipótesis nula. η2: determina la proporción de varianza atribuida a la influencia de la madre (M), a de la dieta en la descendencia (D) o a la interacción entre ambos parámetros (M*D).
En cuanto a la expresión de los genes en el tejido adiposo lumbar tras el test de Bonferroni a posteriori (Figura 11) se encontró que, de nuevo, tanto DIO1 como UCP3 presentaban perfiles muy similares. Así, se hallaron diferencias estadísticamente significativas únicamente en los descendientes de madres-control, donde observamos que los animales que recibieron fructosa (CF) presentaron un aumento muy marcado en la expresión de DIO1 comparado con los que consumieron agua (CC) y los que consumieron tagatosa (CT); A su vez, se encontraron diferencias estadísticamente significativas entre ese grupo CF y los que consumieron fructosa y eran hijos de madres-fructosa (FF). Se encontraron resultados similares en la expresión de UCP3 (Figura 11) mostrando un aumento en los descendientes de madres control suplementados con fructosa (CF) frente a los otros dos grupos (CC o CT) y, por otro lado, también se observaron diferencias significativas de ese grupo CF con respecto a los animales que también tomaron fructosa, pero eran hijos de madres-fructosa (FF).
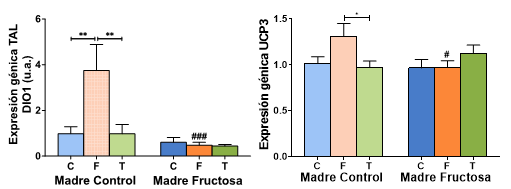
Figura 11. Expresión génica de DIO1, THRa y UCP3 en TAL de machos F1 tras los 21 días de tratamiento (barra azul: agua-C; barra naranja: fructosa-F; barra verde: tagatosa-T). Las barras más claras pertenecen a los machos F1 descendientes de madres control (recibieron agua sin aditivos durante la gestación), mientras que las oscuras pertenecen a machos F1 descendientes de madres fructosa (recibieron fructosa como tratamiento en el agua de bebida durante la gestación). Los datos presentados son las medias ± error estándar (S.E.). El símbolo * indica diferencias estadísticamente significativas entre dietas, y el # entre la ingesta materna.
El consumo de tagatosa en tejido adiposo lumbar no produce un efecto significativo en la expresión génica de las proteínas estudiadas. Por otro lado, la fructosa produce un aumento de la expresión de DIO1 con respecto a la tagatosa y al control en descendientes de madres control, sin embargo, el efecto no se observa en los descendientes de madre fructosa (FF). Se hace patente un efecto significativo en la expresión de UCP3 de la misma forma que en DIO1, la fructosa produce un aumento en la expresión de UCP3 sólo cuando la madre es control. Por tanto, en TAL, al igual que vimos en íleon (pero no en hígado), la fructosa produce en la descendencia cambios tanto en la expresión de DIO1 como de las UCPs, que son dependientes de la ingesta materna y que no están relacionados con los niveles de hormona en plasma.
4.2. Efectos de la fructosa vs “Western diet” (fructosa + colesterol)
4.2.a. T4 libre en plasma
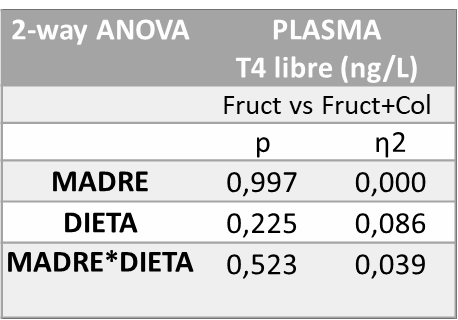
En cuanto a los efectos producidos por la fructosa vs. fructosa+colesterol en la descendencia, los resultados del análisis estadístico de varianza de doble vía (ANOVA) de la Tabla 6 muestran que no se observó ninguna diferencia estadísticamente significativa en los niveles de T4 libre en plasma entre ninguno de los grupos del estudio.
Tabla 6. ANOVA de doble vía T4 libre en plasma fructosa vs fructosa+colesterol. P valor: indica la probabilidad de obtener un valor tan o más extremo al observado, partiendo de la suposición de la igualdad de efecto que marca la hipótesis nula. η2: determina la proporción de varianza atribuida a la influencia de la madre (M), a de la dieta en la descendencia (D) o a la interacción entre ambos parámetros (M*D).
Tras la posterior corrección de los resultados por el test de Bonferroni (Figura 12) tampoco se encontraron diferencias estadísticamente significativas en cuanto a la concentración (ng/L) de T4 libre en plasma en estos grupos.
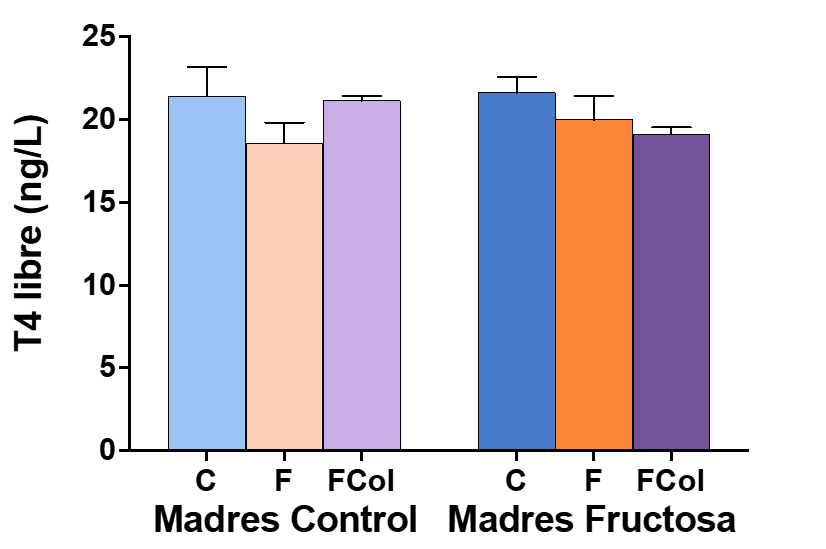
Figura 12. Concentración ng/L T4 libre en plasma de machos F1 tras los 21 días de tratamiento (barra azul: agua-C; barra naranja: fructosa-F; barra morada: fructosa+colesterol-W)). Las barras más claras pertenecen a los machos F1 descendientes de madres control (recibieron agua sin aditivos durante la gestación), mientras que las oscuras pertenecen a machos F1 descendientes de madres fructosa (recibieron fructosa como tratamiento en el agua de bebida durante la gestación). Los datos presentados son las medias ± error estándar (S.E.). El símbolo * indica diferencias estadísticamente significativas entre dietas, y el # entre la ingesta materna.
4.2.b. Hígado
En cuanto al estudio de los efectos de la fructosa comparado con la administración conjunta de fructosa con colesterol en la expresión de los tres genes, (ANOVA de doble vía) (Tabla 7), se observaron diferencias estadísticamente significativas en la expresión de DIO1 por la influencia de la dieta recibida por los machos (D, p=0,049) y por la interacción de ambos tratamientos (M*D, p=0,001).
Tabla 7. ANOVA de doble vía expresión génica DIO1, THRa y UCP2 en hígado. P valor: indica la probabilidad de obtener un valor tan o más extremo al observado, partiendo de la suposición de la igualdad de efecto que marca la hipótesis nula. η2: determina la proporción de varianza atribuida a la influencia de la madre (M), a de la dieta en la descendencia (D) o a la interacción entre ambos parámetros (M*D).
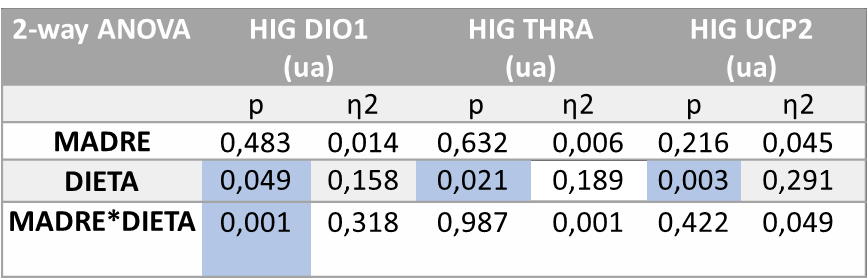
Estas diferencias también se encontraron en la expresión de THRa en el hígado, influenciada por el tratamiento recibido por la descendencia (D, p=0,021) y en la expresión de UCP2 en el hígado, en función de la dieta de la descendencia (D, p=0,003).
Los resultados de la expresión de los genes medidos en hígado tras aplicar a posteriori la corrección de Bonferroni mostraron, como se observa en la Figura 13, que existe un aumento significativo en la expresión de DIO1 en los animales descendientes de madres control que consumieron fructosa con colesterol (CFCol) frente a los que consumieron solamente fructosa (CF) y los que tomaron agua sin aditivos (CC). Sin embargo, dicho efecto no se observó en los descendientes de madres-fructosa y por eso encontramos una diferencia significativa entre dicho grupo CFCol y el correspondiente FFCol. Así pues, este efecto en la expresión de DIO1 era debido a la presencia del colesterol en la dieta y dependiente de la dieta materna. Y, al contrario de lo observado con la tagatosa (Figura 9), no está relacionado con los niveles de T4 en plasma.
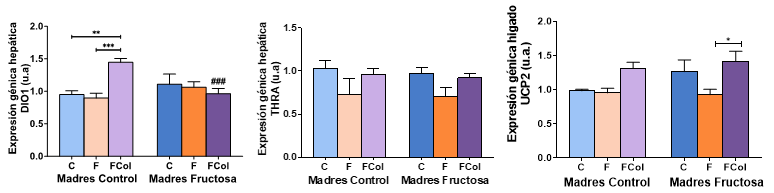
Figura 13. Expresión génica de DIO1, THRa y UCP2 en hígado de machos F1 tras los 21 días de tratamiento (barra azul: agua-C; barra naranja: fructosa-F; barra morada: fructosa+colesterol-FCol). Las barras más claras pertenecen a los machos F1 descendientes de madres control (recibieron agua sin aditivos durante la gestación), mientras que las oscuras pertenecen a machos F1 descendientes de madres fructosa (recibieron fructosa como tratamiento en el agua de bebida durante la gestación). Los datos presentados son las medias ± error estándar (S.E.). El símbolo * indica diferencias estadísticamente significativas entre dietas, y el # entre la ingesta materna.
En cuanto a la expresión de THRa, la disminución debida a la ingesta de fructosa que habíamos encontrado en descendientes de madres-fructosa y, sobre todo, en los de madres control (Figura 9), desaparece cuando dicha fructosa se combina con colesterol en la dieta (Figura 13). Y con respecto a la expresión de UCP2 (Figura 13) se encontró algo curioso, el perfil de los hijos de madres control se asemeja al encontrado para DIO1, mientras que en los machos provenientes de madres-fructosa es más similar al descrito para THRa. Así pues, la expresión del gen diana (UCP2) en los descendientes de madres control experimenta un aumento provocado por la ingesta de la dieta occidental (CFCol) con respecto a los restantes grupos (CC y CF), pero ese efecto no se observó en los descendientes de madres-fructosa. Sin embargo, la disminución observada en la expresión de UCP2 por la ingesta de fructosa en los hijos de madres-fructosa (FF) desaparece tras la adición de colesterol a la dieta (FFCol) alcanzando valores similares a los controles (FC).
4.2.c. Íleon
Los resultados del análisis de varianza ANOVA de doble vía en cuanto a la expresión de los genes en el íleon mostraron que existían diferencias estadísticamente significativas relativas a la expresión de DIO1 en función del tratamiento recibido por la madre (M, p=0,006) e influenciado también por la dieta de la descendencia (D, p=0,025). Por otro lado, también se observan diferencias estadísticamente significativas en UCP2 (p=0.003) influenciadas por el tratamiento recibido por la madre (M) (Tabla 8).
Tabla 8.
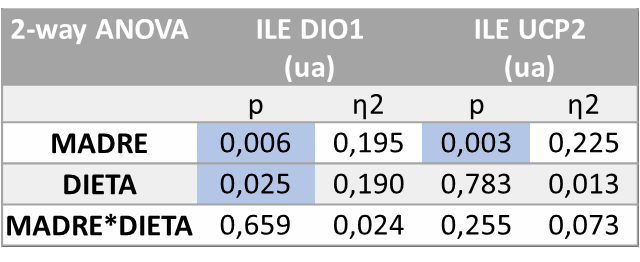
Tras la corrección a posteriori mediante el test de Bonferroni (Figura 14), en cuanto a la expresión génica de DIO1 se observa que la adición de colesterol a la dieta con fructosa acentúa la tendencia a la disminución observada cuando sólo estaba presente la fructosa y, de nuevo, es un efecto más evidente en los hijos de madres-fructosa. Con todo, dicha disminución no llega a alcanzar diferencias significativas en comparación con el resto de los grupos.
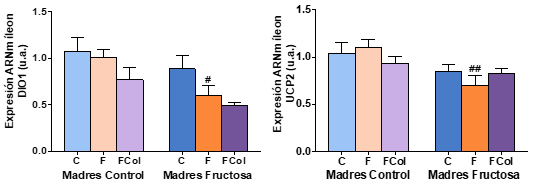
Figura 14. Expresión génica de DIO1 y UCP2 en íleon de machos F1 tras los 21 días de tratamiento (barra azul: agua-C; barra naranja: fructosa-F; barra morada:fructosa+colesterol-FCol). Las barras más claras pertenecen a los machos F1 descendientes de madres control (recibieron agua sin aditivos durante la gestación), mientras que las oscuras pertenecen a machos F1 descendientes de madres fructosa (recibieron fructosa como tratamiento en el agua de bebida durante la gestación). Los datos presentados son las medias ± error estándar (S.E.). El símbolo * indica diferencias estadísticamente significativas entre dietas, y el # entre la ingesta materna.
Sin embargo, en este caso y al contrario de lo observado con la dieta de fructosa (CF vs FF), la tendencia que la Western diet (FFCol) ejerce en la expresión de DIO no se ve reflejada en la expresión de UCP2, ya que no se encuentra modificada.
4.2.1. Tejido Adiposo Lumbar (TAL)
Por último, se estudió la expresión en tejido adiposo lumbar. En este caso, tras la ANOVA de doble vía (Tabla 9) se encontraron diferencias estadísticamente significativas en la expresión de DIO1 influenciada por la dieta recibida por la madre durante la gestación (M, p=0,006), por la dieta recibida por la descendencia (D, p=0,003) y por la interacción entre ambos tratamientos (M*D, p=0,004).
Tabla 9. ANOVA de doble vía expresión génica DIO1, THRa y UCP3 en TAL. P valor: indica la probabilidad de obtener un valor tan o más extremo al observado, partiendo de la suposición de la igualdad de efecto que marca la hipótesis nula. η2: determina la proporción de varianza atribuida a la influencia de la madre (M), a de la dieta en la descendencia (D) o a la interacción entre ambos parámetros (M*D).
La corrección a posteriori por Bonferroni se muestra en la Figura 15. Curiosamente, el aumento significativo en la expresión de DIO1 en este tejido, en el grupo de hijos de madres control que consumieron fructosa en el agua de bebida (CF) comparado con el grupo control (CC), no se observó cuando se añadió colesterol asociado a fructosa (CFCol). Esa adición de colesterol a la dieta con fructosa tampoco fue capaz de inducir cambios en la expresión de DIO1 en los hijos de madres fructosa (FFCol), al igual que había ocurrido con los que sólo tomaron fructosa (FF). Se encontraron resultados similares en la expresión génica de UCP3. La adición de colesterol a la dieta anuló el efecto inductor de la fructosa (la cual alcanzaba a ser cuasi-significativa, p = 0,051, CF frente a CC) en los descendientes de madres control (Figura 15).
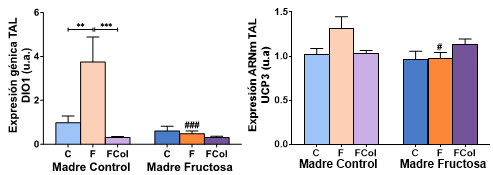
Figura 15. Expresión génica de DIO1 y UCP3 en TAL de machos F1 tras los 21 días de tratamiento (barra azul: agua-C; barra naranja: fructosa-F; barra morada: fructosa+colesterol-FCol). Las barras más claras pertenecen a los machos F1 descendientes de madres control (recibieron agua sin aditivos durante la gestación), mientras que las oscuras pertenecen a machos F1 descendientes de madres fructosa (recibieron fructosa como tratamiento en el agua de bebida durante la gestación). Los datos presentados son las medias ± error estándar (S.E.). El símbolo * indica diferencias estadísticamente significativas entre dietas, y el # entre la ingesta materna.
Así pues, se observa que el efecto de la ingesta de fructosa en descendientes de madres control aumenta la expresión génica de DIO1 y de UCP3, efecto que no se observó cuando la madre se trató con fructosa durante la gestación. La ingesta conjunta de fructosa y colesterol no produjo el mismo efecto que el consumo de fructosa sola.
Conflictos de Interés
Los autores declaran no tener ningún tipo de conflicto de interés, aceptando las bases de este premio.
5. CONCLUSIONES
Las conclusiones del presente estudio en relación a los resultados obtenidos son:
- Los niveles de T4 libre en plasma se ven incrementados por el consumo de tagatosa en los descendientes de madres fructosa, no observándose dicho efecto ni con la ingesta de fructosa ni con la combinación de ésta con colesterol. Además, este es un efecto claramente dependiente de la dieta materna.
- Los cambios observados en la T4 libre sólo afectaron a la expresión de DIO1 en hígado y no en el resto de los tejidos.
- La expresión del gen diana para hormonas tiroideas estudiado en el presente trabajo (UCP2) presentó un perfil en hígado más similar al observado para THRa que al de T4 o al de DIO1, lo que confirma la posibilidad de que estos receptores puedan regular la expresión génica también en ausencia de ligando.
- Tanto en íleon como en tejido adiposo lumbar, los perfiles de expresión encontrados para DIO1 y UCP son similares y experimentan cambios inducidos por el consumo de fructosa, observándose un aumento de ambas proteínas en el tejido adiposo lumbar de los descendientes de madres control. Nuevamente estos efectos fueron dependientes de la ingesta materna.
- Los efectos observados por el consumo de fructosa en hígado (para THRa y UCP2) y en TAL (para DIO1 y UCP3), no se encontraron tras la adición de colesterol en la dieta. Sin embargo, los efectos de fructosa en íleon (para DIO1) sí que se vieron potenciados por la presencia de colesterol.
- Con todos estos resultados, podemos confirmar que el consumo de fructosa materno durante la gestación afecta al metabolismo de las hormonas tiroideas de la descendencia, tanto en respuesta a una dieta rica en fructosa como en forma de “Western diet” (fructosa y colesterol) poniendo de manifiesto un claro efecto de programación fetal.
- Este trabajo pretende alertar a la población, en especial a las mujeres embarazadas del papel relevante que ejerce la nutrición durante este periodo, dado que puede tener consecuencias negativas, no solo en la propia salud de la madre, sino también en la salud de sus hijos en la etapa adulta. El papel del farmacéutico es esencial y clave en este proceso al poder ejercer su consejo dietético en la oficina de farmacia insistiendo a las embarazadas en la necesidad de adquirir hábitos saludables asociados a un consumo limitado de azúcares y de alimentos procesados, para evitar las consecuencias indeseables de su ingesta en la salud de sus hijos.
Índice de abreviaturas
- DIO: desyodasa (deiodinase)
- UCP: proteína desacoplante (uncoupling protein)
- THR: receptor nuclear de hormonas tiroideas (thyroid hormone nuclear receptor)
- TAL: tejido adiposo lumbar (lumbar adipose tissue)
Agradecimientos
Los autores agradecen a J.M. Garrido y su equipo su ayuda en el manejo de los animales. Este trabajo ha sido financiado por el Ministerio de Ciencia e Innovación (MICIN) (PID2020-118054RB-I00/MCIN/AEI/10.13039/501100011033). Elena Fauste disfrutó de una beca FPU del MICIN.
6. REFERENCIAS
- Lee W., Wu K.L.H., Leu S.and Tain Y.L. (2018). “Translational insights on developmental origins of metabolic syndrome: Focus on fructose consumption”. Biomedical Journal 41(2). 96-101. doi:10.1016/j.bj.2018.02.006
- Girirajan, S., Campbell, C., and Eichler, E. (2011). “Added sugars intake across the distribution of US children and adult consumers: 1977–2012”. Physiology & Behavior, 176(5), 139–148. doi:10.1016/j.jand.2016.06.003.
- Hanover, L. M., and White, J. S. (1993). “Manufacturing, composition, and applications of fructose”. The American journal of clinical nutrition, 58(5 Suppl), 724S–732S. doi:10.1093/ajcn/58.5.724S
- Dornas, W. C., de Lima, W. G., Pedrosa, M. L., and Silva, M. E. (2015). “Health implications of high-fructose intake and current research”. Advances in Nutrition, 6(6), 729–737. doi:10.3945/an.114.008144
- Bray, G.A., Nielsen, S.J. and Popkin, B.M. (2004) “Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity,” American Journal of Clinical Nutrition, 79(4), pp. 537–543. doi:10.1093/ajcn/79.4.537.
- Myles, I. A. (2014). “Fast food fever: reviewing the impacts of the Western diet on immunity”. Nutr J, 13, 61. doi:10.1186/1475-2891-13-61
- Cena, H., and Calder, P. C. (2020). “Defining a Healthy Diet: Evidence for The Role of Contemporary Dietary Patterns in Health and Disease”. Nutrients, 12(2). doi:10.3390/nu12020334
- Taskinen, M. R., Packard, C. J., and Borén, J. (2019a). “Dietary fructose and the metabolic syndrome”. Nutrients, 11(9), 1–16. doi:10.3390/nu11091987
- Younossi, Z. M. (2019). “Non-alcoholic fatty liver disease – A global public health perspective”. Journal of Hepatology, 70(3), 531–544. doi:10.1016/j.jhep.2018.10.033
- Mellor, K. M., Ritchie, R. H., Davidoff, A. J., and Delbridge, L. M. D. (2010). “Elevated dietary sugar and the heart: experimental models and myocardial remodeling”. Can. J. Physiol. Pharmacol. 88, 525–540. doi:10.1139/Y10-005
- Durante, M., Sgambellone, S., Lucarini, L., Failli, P., Laurino, A., Collotta, D., Provensi, G., Masini, E., and Collino, M. (2021). “D-Tagatose Feeding Reduces the Risk of Sugar-Induced Exacerbation of Myocardial I/R Injury When Compared to Its Isomer Fructose”. Frontiers in Molecular Biosciences, 8(April), 1–12. doi:10.3389/fmolb.2021.650962
- Chan, A. M. L., Ng, A. M. H., Mohd Yunus, M. H., Idrus, R. B. H., Law, J. X., Yazid, M. D., Chin, K. Y., Shamsuddin, S. A., and Lokanathan, Y. (2021). “Recent developments in rodent models of high-fructose diet-induced metabolic syndrome: A systematic review”. Nutrients, 13(8), 1–21. doi:10.3390/nu13082497
- Agodi, A. et al. (2018) “Association of dietary patterns with metabolic syndrome: Results from the kardiovize brno 2030 study,” Nutrients, 10(7). doi:10.3390/nu10070898.
- Williams, J. (2013). “The Effect of D-Tagatose on Fructose Absorption in a Rat Model”. Journal of Developing Drugs, 02(03). doi:10.4172/2329-6631.1000111
- Levin G. V. (2002). “Tagatose, the new GRAS sweetener and health product”. Journal of medicinal food, 5(1), 23–36. doi:10.1089/109662002753723197
- Levin, G. V., Zehner, L. R., Saunders, J. P. and Beadle, I. R. (1995). “Sugar substitutes, their energy values, bulk characteristics andpotential health benefits”. Am. Clin. Nutr. 62 (suppl. 1): 1161S1168S
- Vastenavond, Christian & Bertelsen, Hans & Hansen, S.J. & Laursen, R.S. & Saunders, James & Eriknauer, K. (2016). “Tagatose (D-tagatose)”.
- Donner, T. W., Wilber, J. F., and Ostrowski, D. (1999). “D-tagatose, a novel hexose: acute effects on carbohydrate tolerance in subjects with and without type 2 diabetes”. Diabetes, obesity & metabolism, 1(5), 285–291. doi:10.1046/j.1463-1326.1999.00039.x
- Kim P. (2004). “Current studies on biological tagatose production using L- arabinose isomerase: a review and future perspective”. Applied microbiology and biotechnology, 65(3), 243–249. doi:10.1007/s00253-004-1665-8
- Collotta, D., Lucarini, L., Chiazza, F., Cento, A. S., Durante, M., Sgambellone, S., Chini, J., Baratta, F., Aragno, M., Mastrocola, R., Masini, E., and Collino, M. (2018). “Reduced susceptibility to sugar-induced metabolic derangements and impairments of myocardial redox signaling in mice chronically fed with D- tagatose when compared to fructose”. Oxidative Medicine and Cellular Longevity, 2018. doi:10.1155/2018/5042428.
- Herrera, E.; Ramos M.P.; Roca P.; and Viana, M.: “Bioquímica Básica”. Ed. Elsevier, 2014.
- Lehninger, A.L.; Nelson, D.L.; and Cox M.M.: “Principios de Bioquímica”. 5ª Edición. Ed. Omega, S.A. Barcelona. 2009.
- Sun, S. z., and Empie, M. W. (2012). “Fructose metabolism in humans–what isotopic tracer studies tell us.: University at Albany Libraries eDiscover Service”. Nutrition and Metabolism, 9(89), 1–15.
- Douard, V., and Ferraris, R. P. (2008). “Regulation of the fructose transporter GLUT5 in health and disease”. American Journal of Physiology – Endocrinology and Metabolism, 295(2). doi:10.1152/ajpendo.90245.2008
- Mráček, T., Drahota, Z., and Houštěk, J. (2013). “The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues”. Biochimica et Biophysica Acta – Bioenergetics, 1827(3), 401–410. doi:10.1016/j.bbabio.2012.11.014
- Krause, N., and Wegner, A. (2020). “Fructose Metabolism in Cancer”. Cells, 9(12). doi:10.3390/cells9122635
- Lu, Y., Levin, G. V., and Donner, T. W. (2008). “Tagatose, a new antidiabetic and obesity control drug”. Diabetes, obesity and metabolism, 10(2), 109-134. doi:10.1111/j.1463-1326.2007.00799.x
- Mendoza A., and Hollenberg A.N. (2017). “New insights into thyroid hormone action”. Pharmacology & Therapeutics, 173, 135-145. doi:10.1016/j.pharmthera.2017.02.012.
- Brent, G. A. (2012). “Mechanisms of thyroid hormone action”. In Journal of Clinical Investigation (Vol. 122, Issue 9, pp. 3035–3043). doi:10.1172/JCI60047.
- Mullur, R., Liu, Y. Y., and Brent, G. A. (2014). “Thyroid hormone regulation of metabolism”. Physiological reviews, 94(2), 355–382. doi:10.1152/physrev.00030.2013
- Williams, G. R., and Bassett, J. H. D. (2011). “Local control of thyroid hormone action: Role of type 2 deiodinase”. In Journal of Endocrinology (Vol. 209, Issue 3, pp. 261–272). doi:10.1530/JOE-10-0448
- Visser, W. E., Friesema, E. C., Jansen, J., and Visser, T. J. (2007). “Thyroid hormone transport by monocarboxylate transporters. Best practice & research”. Clinical endocrinology & metabolism, 21(2), 223–236. doi:10.1016/j.beem.2007.03.008
- van der Deure, W. M., Peeters, R. P., and Visser, T. J. (2010). ”Molecular aspects of thyroid hormone transporters, including MCT8, MCT10, and OATPs, and the effects of genetic variation in these transporters”. Journal of molecular endocrinology, 44(1), 1–11. doi:10.1677/JME-09-0042
- Benjamin Chun-Kit Tong (2017) “乳鼠心肌提取 HHS Public Access,” Physiology & behavior, 176(5), pp. 139–148. doi:10.1016/j.pharmthera.2017.02.012.New.
- Gereben, B., Zavacki, A. M., Ribich, S., Kim, B. W., Huang, S. A., Simonides, W. S., Zeöld, A., and Bianco, A. C. (2008). “Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling”. Endocrine reviews, 29(7), 898–938. doi:10.1210/er.2008-0019
- Brand, M.D. and Esteves, T.C. (2005) “Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3,” Cell Metabolism, 2(2), pp. 85–93. doi:10.1016/j.cmet.2005.06.002.
- Aguilar Salinas, C.A. (2002) “Las proteínas desacoplantes en la patogenia de la obesidad,” Revista de Endocrinología y Nutrición, 10(3), pp. 165–170. Available at: http://www.medigraphic.com/pdfs/endoc/er-2002/er023j.pdf.
- Bruinstroop, E., Zhou, J., Tripathi, M., Yau, W. W., Boelen, A., Singh, B. K., and Yen, P. M. (2021a). “Early induction of hepatic deiodinase type 1 inhibits hepatosteatosis during NAFLD progression”. Molecular Metabolism, 53(June), 101266. doi:10.1016/j.molmet.2021.101266
- Hales, C.N., and Barker, D.J.P. “Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis”. Diabetologia 35, 595–601 (1992). doi:10.1007/BF00400248
- Wahlqvist, M. L., Krawetz, S. A., Rizzo, N. S., Dominguez-Bello, M. G., Szymanski, L. M., Barkin, S., Yatkine, A., Waterland, R. A., Mennella, J. A., Desai, M., Ross, M. G., Krebs, N. F., Young, B. E., Wardle, J., Wrann, C. D., and Kral, J. G. (2015). “Early-life influences on obesity: From preconception to adolescence”. Annals of the New York Academy of Sciences, 1347(1), 1–28. doi:10.1111/nyas.12778
- Kwon, E. J., and Kim, Y. J. (2017). “What is foetal programming?: A lifetime health is under the control of in utero health”. Obstetrics and Gynecology Science, 60(6), 506–519. doi:10.5468/ogs.2017.60.6.506
- Fauste, E., Panadero, M. I., Donis, C., Otero, P., and Bocos, C. (2021). “Pregnancy is enough to provoke deleterious effects in descendants of fructose‐fed mothers and their fetuses”. Nutrients, 13(10). doi:10.3390/nu13103667
- Portha, B., Grandjean, V., and Movassat, J. (2019). “Mother or father: Who is in the front line? Mechanisms underlying the non-genomic transmission of obesity/diabetes via the maternal or the paternal line”. Nutrients, 11(2). doi:10.3390/nu11020233
- Herráez, Á. (2010). Análisis de biomarcadores empleando biochips y matrices de biomoléculas. Monografías de la Real Academia Nacional de Farmacia.
- Pfaffl M.W., (2001) “A new mathematical model for relative quantification in real-time RT-PCR”. Nucleic Acids Research, 29 (9). e45-e45.
- Judith Hallfrisch, Ph.D., Sheldon Reiser, Ph. D., and Elizabeth S. Prather, Ph.D., J.J. Canary, M. D. (1983). “Relationships of glucoregulatory hormones in normal and hyperinsulinemic men consuming fructose”. 3(c), 119–128.
- Jong, M. de, Docter, R., Bernard, B. F., M Van Der Heijden, J. T., van Toor, H., Krenning, E. P., Hennemann, G., Jong, D., Dotter, R., M van der Heijden, J. T., and van Toor, H. (n.d.). “T4 uptake into the perfused rat liver and liver T4 uptake in humans are inhibited by fructose”. www.physiology.org/journal/ajpendo
- Solmonson, A., & Mills, E. M. (2016). Uncoupling Proteins and the Molecular Mechanisms of Thyroid Thermogenesis. Endocrinology, 157(2), 455–462. https://doi.org/10.1210/en.20151803
