(A) TRACTO ALIMENTARIO Y METABOLISMO: Hiperfenilalaninemia: Sepiapterina (Sephience®; UE). (B) SANGRE Y SISTEMA HEMATOPOYÉTICO: Trasplante alogénico de células madre hematopoyéticas: Dorocubicel + Células de cordón umbilical no expandidas (Zemcelpro®; UE). Enfermedad de la orina con olor a jarabe de arce: Aminoácidos de cadena no ramificada (Maapliv®; UE).(C) SISTEMA CARDIOVASCULAR: Nefropatía por Inmunoglobulina A: Atrasentan (Vanrafia®; USA). (D) DERMATOLOGÍA: Epidermolisis ampollosa distrófica recesiva: Prademagene Zamikeracel (Zebaskyn®; USA). (J) ANTIINFECCIOSOS SISTÉMICOS: Prevención de la infección respiratoria por VRS: Clesrovimab (Enflonsia®; USA). (L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES: Carcinoma nasofaríngeo: Penpulimab (Penpulimab®; USA). Mieloma múltiple: Linvoseltamab (Lynozyfic®; UE). Miastenia grave: Nipocalimab (Imaavy®; USA). Cáncer de ovario: Avutometinib + Defactinib (Avmapki Fakzynja Co-Pack®; USA). Cáncer de pulmón: Telisotuzumab Vedotina (Emrelis®; USA). Cáncer de pulmón: Taletractinib (Ibtrozi®; USA). Enfermedad ocular del tiroides: Teprotumumab (Tepezza®; UE/USA). Mielosupresión inducida por radioterapia: Sargramostim (Imreplys®; UE/USA). (N) SISTEMA NERVIOSO: Amiloidosis por transtiretina: Diflunisal (Attrogy®; UE). (S) ÓRGANOS SENSORIALES: Telangiectasia macular idiopática tipo 2: Revakinagene Taroretcel (Encelto®; USA). Enfermedad del ojo seco: Acoltremon (Tryptyr®; USA).
ABSTRACT:
(A) GASTROINTESTINAL TRACT AND METABOLISM: Hyperphenylalaninemia: Sepiapterin (Sephience®; EU). (B) BLOOD AND HEMATOPOIETIC SYSTEM: Allogeneic hematopoietic stem cell transplantation: Dorocubicel + Unexpanded umbilical cord blood cells (Zemcelpro®; EU). Maple syrup urine disease: Branched-chain amino acids (Maapliv®; EU).(C) CARDIOVASCULAR SYSTEM: Immunoglobulin A Nephropathy: Atrasentan (Vanrafia®; USA). (D) DERMATOLOGY: Recessive dystrophic epidermolysis bullosa: Prademagene Zamikeracel (Zebaskyn®; USA). (J) SYSTEMIC ANTIBIOTICS: Prevention of respiratory infection by RSV: Clesrovimab (Enflonsia®; USA). (L) ANTINEOPLASTIC AND IMMUNOMODULATORY AGENTS: Nasopharyngeal carcinoma: Penpulimab (Penpulimab®; USA). Multiple myeloma: Linvoseltamab (Lynozyfic®; EU). Myasthenia gravis: Nipocalimab (Imaavy®; USA). Ovarian cancer: Avutometinib + Defactinib (Avmapki Fakzynja Co-Pack®; USA). Lung cancer: Telisotuzumab Vedotina (Emrelis®; USA). Lung cancer: Taletractinib (Ibtrozi®; USA). Thyroid eye disease: Teprotumumab (Tepezza®; EU/USA). Radiotherapy-induced myelosuppression: Sargramostim (Imreplys®; EU/USA). (N) NERVOUS SYSTEM: Transthyretin amyloidosis: Diflunisal (Attrogy®; EU). (S) SENSORY ORGANS: Idiopathic Macular Telangiectasia Type 2: Revakinagene Taroretcel (Encelto®; USA). Dry eye disease: Acoltremon (Tryptyr®; USA).
(A) TRACTO ALIMENTARIO Y METABOLISMO
SEPIAPTERINA (SEPHIENCE®) PTC (UE)
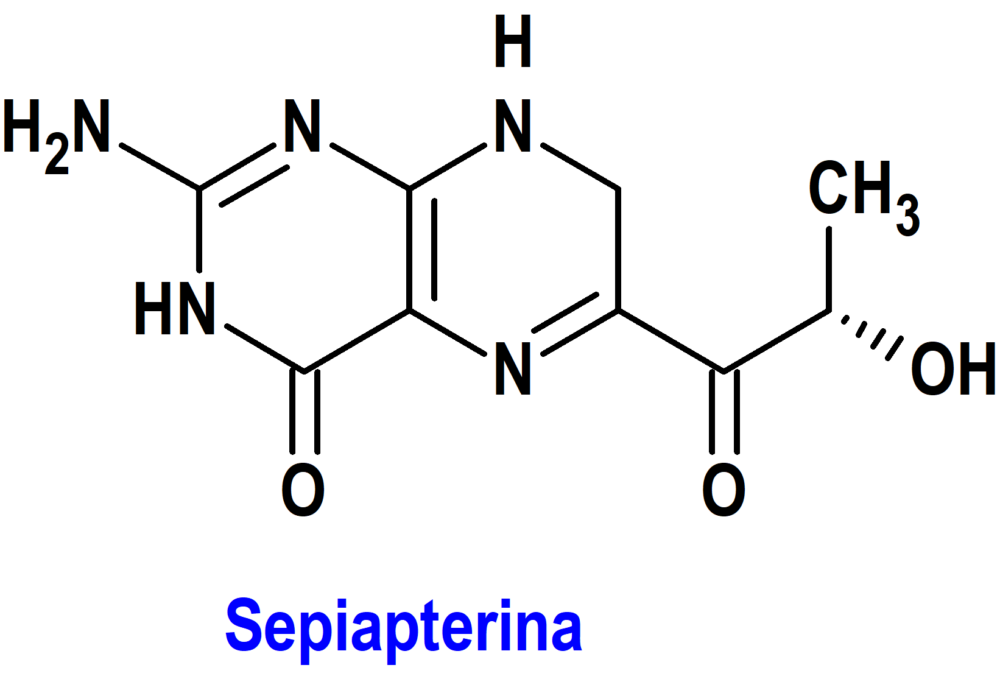
Indicación: Tratamiento de la hiperfenilalaninemia en pacientes adultos y pediátricos con fenilcetonuria (PKU).
Tipo: Medicamento sintético estándar constituido por la 2-amino-6-[(2S)-2-hidroxipropanoil]-7,8-dihidro-3H-pteridin-4-ona. Autorizado en la Unión Europea el 19 de junio de 2025 como medicamento huérfano (Orphan drug); no disponible en Estados Unidos.
Mecanismo: Es un precursor natural del cofactor enzimático BH4 (tetrahidrobiopterina), un cofactor crítico de la fenilalanina hidroxilasa (PAH). La sepiapterina actúa como una doble chaperona farmacológica (sepiapterina y BH4, cada una con su propia afinidad de unión a la variante de PAH), incluyendo variantes de PAH que se encuentran comúnmente en la fenilcetonuria y que se sabe que son insensibles a BH4, para mejorar la actividad de la enzima PAH defectuosa, logrando una alta concentración intracelular de BH4. Al mejorar la estabilidad conformacional de la enzima de PAH mal plegada y aumentar las concentraciones intracelulares de BH4, la sepiapterina es capaz de reducir eficazmente los niveles de Phe en sangre.
Eficacia clínica: Tres estudios clínicos en pacientes con fenilcetonuria (PKU). El estudio 1 fue un estudio clínico de 2 partes, a nivel mundial, doble ciego, aleatorizado y controlado con placebo de 157 pacientes de todas las edades con PKU. En la parte 1 del estudio se evaluó la respuesta a sepiapterina, con 14 días de tratamiento sin enmascaramiento con sepiapterina seguidos de un mínimo de 14 días de reposo farmacológico de sepiapterina, tras el cual los pacientes se aleatorizaron por igual para recibir 20 mg/kg/día de sepiapterina durante las semanas 1 y 2, 40 mg/kg/día durante las semanas 3 y 4, 60 mg/kg/día durante las semanas 5 y 6 (n = 56) o placebo (n = 54) durante 6 semanas. La variable principal de eficacia primaria fue el cambio medio en los niveles de fenilalanina (Phe) en sangre desde el inicio hasta las semanas 5 y 6 en el grupo tratado con sepiapterina en comparación con el cambio medio en el grupo de placebo en pacientes que mostraron una reducción ≥30 % en los niveles de Phe en sangre durante la parte 1: -62,8% vs. +1,4%. El estudio 2 fue un estudio clínico de prueba de concepto de fase II, aleatorizado, con doble cruce, abierto y controlado con principio activo de sepiapterina en pacientes con PKU, que consistió en 6 grupos secuenciales de 4 pacientes por grupo, con un total de 24 pacientes, encontrándose que el tratamiento con sepiapterina (60 mg/kg/día) produjo una disminución significativa de la concentración de Phe en sangre con respecto al inicio. El estudio 3 es un estudio clínico de fase III, multicéntrico y abierto en curso para evaluar la seguridad y la tolerancia a la Phe en la dieta durante el tratamiento a largo plazo con sepiapterina en pacientes con PKU; los datos provisionales indican que la administración diaria de sepiapterina se asocia a un aumento aproximado de 2,3 veces en el consumo medio diario de Phe (27,6 mg/kg/día al inicio frente a 62,5 mg/kg/día en la semana 26) al tiempo que se mantienen niveles de Phe <360 μmol/, donde la mayoría de los sujetos alcanzaron al menos una reducción del 15% (77%) o del 30 % (67%) en los niveles de Phe en sangre. Eventos adversos: Los más comunes son infección de las vías respiratorias superiores (20%), dolor de cabeza (15%), diarrea (15%), dolor abdominal (12%), heces con cambio de color (4,5%) y hipofenilalaninemia (2,7%).
(B) SANGRE Y SISTEMA HEMATOPOYÉTICO
DOROCUBICEL + CELULAS CD34- DE CORDÓN UMBILICAL (ZEMCELPRO®) CORDEX (UE)
Indicación: Tratamiento de adultos con neoplasias hematológicas. Puede utilizarse en pacientes que requieren un trasplante alogénico de células madre hematopoyéticas (alo-TCMH) tras un acondicionamiento mieloablativo (quimioterapia o radioterapia) y para quienes no se dispone de otro tipo de células de un donante adecuado. Tipo: Medicamento de terapia avanzada (celular somática), constituido por células madre de la sangre del cordón umbilical de un donante, algunas de las cuales han sido cultivadas y multiplicadas (dorocubicel). Evaluado favorablemente en la Unión Europea (CHMP, EMA) el 19 de junio de 2025 como medicamento huérfano (Orphan drug), con revisión prioritaria (Priority Medicines; PRIME) y condicionalmente (Conditional marketing authorisation); no disponible en Estados Unidos. Mecanismo: Las neoplasias hematológicas son cánceres frecuentemente diagnosticados y la única opción de tratamiento potencialmente curativo para varios de ellos es el alotrasplante de células madre hematopoyéticas, que implica el uso de células madre donadas para reemplazar las células de la médula ósea del receptor y formar nueva médula ósea que produzca células sanguíneas sanas. Al aumentar el número de células, Zemcelpro aumenta la eficacia de las células madre de una pequeña unidad de sangre del cordón umbilical. Eficacia clínica: Dos estudios abiertos de un solo grupo que incluyeron globalmente a 25 pacientes. En total, 21/25 (84%) pacientes lograron el injerto de neutrófilos en una mediana de 20 días, y 17 (68%) pacientes lograron el injerto de plaquetas en una mediana de 40 días. Eventos adversos: Los más comunes son niveles bajos de varios tipos de células sanguíneas y de anticuerpos, hipertensión arterial, infecciones y síndrome de injerto. La enfermedad de injerto contra huésped (EICH) aguda, se reportó hasta 100 días después del trasplante en el 60% de los pacientes, y la EICH crónica, que apareció hasta un año después del trasplante, se reportó en el 13% de los pacientes.
AMINOÁCIDOS DE CADENA NO RAMIFICADA (MAAPLIV®) RECORDATI (UE)
Indicación: Tratamiento de la enfermedad de la orina con olor a jarabe de arce (MSUD) que se presenta con un episodio de descompensación aguda en pacientes desde el nacimiento que no son elegibles para una formulación oral y enteral libre de aminoácidos de cadena ramificada (libre de BCAA). Tipo: Medicamento sintético estándar constituido por una combinación de aminoácidos exenta de aminoácidos de cadena ramificada (AACR) en forma de solución para uso parenteral. Evaluado favorablemente en la Unión Europea (CHMP, EMA) el 23 de mayo de 2025 como medicamento huérfano (Orphan drug), en condiciones excepcionales (Exceptional circumstances); no disponible en Estados Unidos. Mecanismo: En combinación con suplementos de carbohidratos y lípidos, ayuda a normalizar la leucina y previene o revierte el catabolismo proteico y promueve el anabolismo en pacientes con descompensación de MSUD, reduciendo así los niveles de alfa-cetoácidos. Eficacia clínica: Documentado en cinco publicaciones científicas que informan sobre el uso parenteral de soluciones sin BCAA con la misma formulación que Maapliv. Eventos adversos: Leves y poco frecuentes.
(C) SISTEMA CARDIOVASCULAR
ATRASENTAN (VANRAFIA®) NOVARTIS (USA)
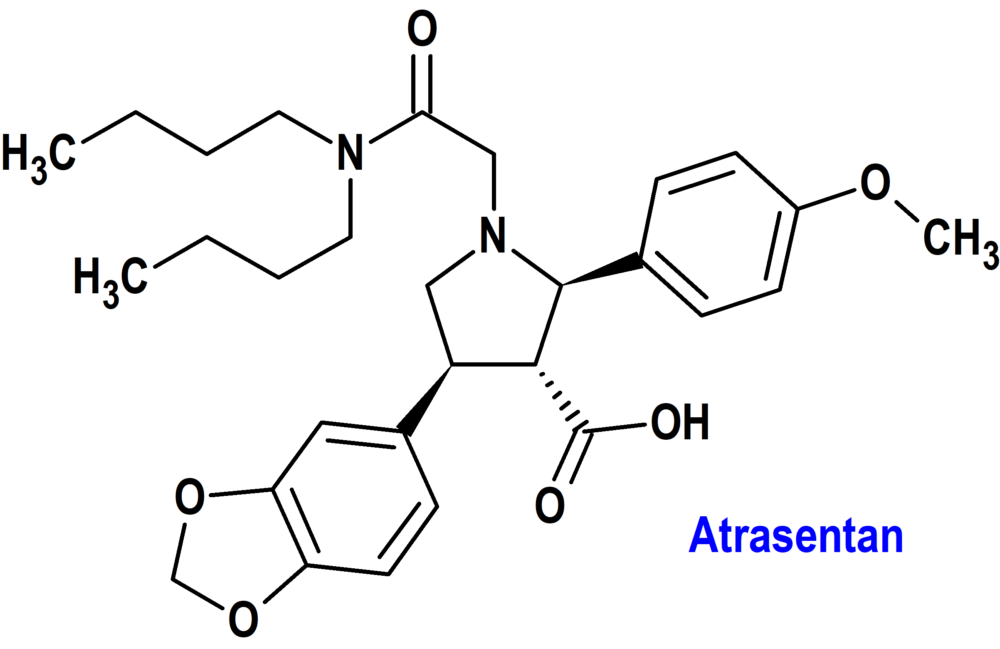
Indicación: Para reducir la proteinuria en adultos con nefropatía primaria por inmunoglobulina A (NIgA) con riesgo de progresión rápida de la enfermedad, generalmente una relación proteína-creatinina en orina (UPCR) ≥1,5 g/g. Tipo: Medicamento sintético estándar constituido por el ácido (2R,3R,4S)-4-(1,3-benzodioxol-5-il)-1-[2-(dibutilamino)-2-oxoetil]-2-(4-metoxifenil)pirrolidina-3-carboxílico. Autorizado (FDA) en Estados Unidos el 2 de abril de 2025 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval), mediante revisión prioritaria (Priority Review); no disponible en la Unión Europea. Mecanismo: Antagonista del receptor de endotelina tipo A (ETA) con una selectividad >1800 veces mayor que para el receptor de endotelina tipo B. Se cree que la endotelina (ET)-1 contribuye a la patogénesis de la NIgA a través del receptor ETA.
Eficacia clínica: Estudio multicéntrico, global, aleatorizado, doble ciego y controlado con placebo en adultos con NIgA primaria confirmada por biopsia, una TFGe ≥ 30 ml/min/1,73 m² y una concentración de proteínas en orina ≥1 g/día con una dosis estable de inhibidor del sistema renina-angiotensina (SRS) máxima tolerada. El estudio incluyó dos cohortes: una principal de 340 pacientes y una exploratoria de 64 pacientes que, al inicio, también recibían una dosis estable de inhibidor del cotransportador de sodio y glucosa tipo 2 (SGLT2i). El análisis de eficacia incluyó a los primeros 270 pacientes de la cohorte principal que acudieron a la consulta de la semana 36. El criterio de valoración principal fue la reducción porcentual del cociente proteína-creatinina en orina (UPCR) en la semana 36 con respecto al inicio: 38% con atrasentan vs. 3% con placebo.
Eventos adversos: Los más comunes (≥2% ) son edema periférico (10%), anemia (6%) y elevación de transaminsas hepáticas (2%).
(D) DERMATOLOGÍA
PRADEMAGENE ZAMIKERACEL (ZEBASKYN®) ABEONA (USA)
Indicación: Tratamiento de heridas en pacientes adultos y pediátricos con epidermólisis ampollosa distrófica recesiva (EBDR).
Tipo: Medicamento de terapia avanzada, constituido por células autólogas aisladas de biopsias cutáneas de pacientes con mutaciones en el gen de la cadena alfa 1 del colágeno tipo VII (COL7A1), transducidas ex vivo con un vector retroviral incompetente para la replicación (RVV) que contiene el gen COL7A1 completo. Las láminas celulares resultantes, modificadas genéticamente, expresan la proteína funcional del colágeno VII (C7). Autorizado en Estados Unidos el 28 de abril de 2025 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review); no disponible en la Unión Europea.
Mecanismo: En pacientes con epidermólisis ampollosa distrófica recesiva (EBDR), ambas copias del gen COL7A1 están mutadas, lo que resulta en la ausencia o niveles bajos de la proteína C7 biológicamente activa, que forma fibrillas de anclaje (FA). La falta de FA altera la conexión entre la epidermis y la dermis, causando fragilidad cutánea y otros signos y síntomas de la EBDR. ZEVASKYN consiste en células del propio paciente, modificadas genéticamente mediante transducción de RVV para expresar el gen COL7A1 y producir la proteína C7. Estas células se transforman en láminas celulares para su aplicación tópica en heridas.
Eficacia clínica: Estudio multicéntrico, aleatorizado y controlado intrapaciente, comparando la aplicación de ZEVASKYN con el tratamiento de referencia en pacientes con heridas asociadas con epidermólisis ampollosa distrófica recesiva (EBDR). Se incluyeron 86 heridas en 11 pacientes, que fueron tratadas con ZEVASKYN o con el tratamiento de referencia. Las variables de eficacia coprincipales fueron: 1) proporción de pares de heridas aleatorizados con al menos un 50% de cicatrización al sexto mes, con confirmación de la cicatrización dos semanas después, según la evaluación del investigador mediante fotografía digital basal (81 vs. 16%); y 2) reducción del dolor, evaluada mediante las diferencias medias en las puntuaciones de dolor notificadas por los pacientes, utilizando la escala Wong-Baker FACES, entre los pares de heridas aleatorizados al sexto mes (-3,07 vs. -0,90). Las variables secundarias fueron la proporción de pares de heridas aleatorizados con cicatrización completa, definida como reepitelización sin drenaje ni erosión, y presencia de solo una pequeña formación de costras desde el inicio al tercer mes (14 vs. 0%) y al sexto mes (16 vs 0%), con confirmación de la cicatrización dos semanas después.
Eventos adversos: Los más comunes (≥5 %) son dolor durante el procedimiento (27%) y prurito (9%).
(J) ANTIINFECCIOSOS SISTÉMICOS
CLESROVIMAB (ENFLONSIA®) MERCK SHARP DOHME (USA)
Indicación: Prevención de la enfermedad de las vías respiratorias inferiores causada por el virus respiratorio sincitial (VRS) en neonatos y bebés que nacen durante o entran en su primera temporada de VRS.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal completamente humano tipo inmunoglobulina G1 kappa (IgG1κ) con un peso molecular de 149 kDa. Autorizado en Estados Unidos el 9 de junio de 2025; no disponible en la Unión Europea.
Mecanismo: Anticuerpo monoclonal recombinante humano de tipo neutralizante con una triple sustitución de aminoácidos YTE (M252Y/S254T/T256E) en la región Fc, lo que aumenta la unión al receptor Fc neonatal, lo que prolonga la semivida sérica. El clesrovimab proporciona inmunidad pasiva, dirigiéndose al dominio extracelular de la proteína de fusión (F) del VRS para prevenir la fusión de las membranas viral y celular, así como la entrada del virus. El clesrovimab se une a un epítopo conservado en el sitio antigénico IV de la proteína F y se une a la glucoproteína F prefusión y posfusión del VRS A, con valores de constante de disociación en equilibrio (KD) de 71 pM y 480 pM, respectivamente.
Eficacia clínica: Dos ensayos clínicos aleatorizados, controlados, doblemente ciegos y multinacionales. El primero fue un ensayo multicéntrico, aleatorizado, doble ciego y controlado con placebo de fase 2b/3, realizado en 22 países de los hemisferios norte y sur, en lactantes prematuros (≥29 a <35 semanas de edad gestacional, EG) y prematuros tardíos y lactantes a término (≥35 semanas EG). El ensayo evaluó la eficacia en la prevención de la enfermedad asociada al VRS en un espectro de gravedad. El criterio de valoración principal fue la incidencia de la infección de las vías respiratorias inferiores con asistencia médica (MALRI) asociada al VRS, caracterizada por tos o dificultad para respirar y que requirió ≥1 indicador de LRI (sibilancias, estertores/crepitaciones) o gravedad (tiraje/retracción de la pared torácica, hipoxemia, taquipnea, deshidratación debido a síntomas respiratorios) hasta 150 días después de la administración. La eficacia para MALRI (≥1 indicador de LRI o gravedad) y la hospitalización se basaron en la reducción del riesgo relativo frente a placebo ajustada por hemisferio en la aleatorización, grupo de edad gestacional y grupo de edad en la aleatorización: MALRI, 60,5% y Hospitalización: 84,3%. El segundo fue un ensayo multicéntrico de fase 3, aleatorizado, parcialmente ciego y controlado con palivizumab, realizado en 27 países de los hemisferios norte y sur, en lactantes prematuros (<29 semanas de EG) o moderados (≥29 a ≤35 semanas de EG), así como en lactantes con enfermedad pulmonar crónica de la prematuridad o cardiopatía congénita de cualquier edad, que presentan un mayor riesgo de enfermedad grave por VRS. La eficacia en lactantes con mayor riesgo de enfermedad grave por VRS, incluyendo lactantes prematuros y lactantes con enfermedad pulmonar crónica de la prematuridad o cardiopatía congénita, se estableció mediante la extrapolación de la eficacia del ensayo anterior a este último, basándose en una exposición farmacocinética similar. Las tasas de incidencia de MALRI asociada al VRS (≥1 indicador de LRI o gravedad) hasta 150 días después de la administración fueron comparables entre clesrovimab y palivizumab (3,6 vs. 2,9%); asimismo, las tasas de incidencia de hospitalización asociada al VRS hasta 150 días después de la administración fueron también comparables (1,3 vs 1,5%).
Eventos adversos: Los más comunes son eritema en el lugar de la inyección (3,8 frente a 3,3 % con placebo), hinchazón en el lugar de la inyección (2,7 frente a 2,6 %), erupción cutánea (2,3 frente a 1,9 %).
(L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES
PENPULIMAB (PENPULIMAB®) AKESO (USA)
Indicación: Tratamiento de primera línea, en combinación con cisplatino o carboplatino y gemcitabina, de adultos con carcinoma nasofaríngeo no queratinizante (NPC) recurrente o metastásico.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal humanizado IgG1 con un peso molecular aproximado de 150 kDa. Autorizado en Estados Unidos el 23 de abril de 2025 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track) y designado como terapia innovadora (Breakthrough Therapy); no disponible en la Unión Europea.
Mecanismo: Penpulimab se une a un epítopo en el sitio de N-glicosilación del PD-1 humano (receptor de muerte programada 1), en el residuo N58, bloqueando su interacción con PD-L1 y PD-L2, liberando así la inhibición de la respuesta inmunitaria mediada por la vía PD-1, incluida la respuesta inmunitaria antitumoral. La unión de los ligandos PD-1, PD-L1 y PD-L2, al receptor PD-1 presente en los linfocitos T inhibe la proliferación de estos y la producción de citocinas. En algunos tumores, se produce una sobreexpresión de los ligandos PD-1, y la señalización a través de esta vía puede contribuir a la inhibición de la vigilancia inmunitaria activa de los linfocitos T en los tumores. Su estructura Fc modificada permite que no interactúe con los receptores Fcγ y, por lo tanto, no induce funciones efectoras de las células inmunitarias (macrófagos) (citotoxicidad celular dependiente de anticuerpos/ADCC, fagocitosis celular dependiente de anticuerpos/ADCP, liberación de citocinas dependiente de anticuerpos/ADCR).
Eficacia clínica: La eficacia de penpulimab asociado con cisplatino o carboplatino y gemcitabina se evaluó en un ensayo aleatorio, doble ciego y multicéntrico en 291 pacientes con cáncer nasofaríngeo no metastásico recurrente o metastásico que no habían recibido terapia sistémica previa para la enfermedad recurrente o metastásica. Los pacientes fueron aleatorizados (1:1) para recibir penpulimab con cisplatino o carboplatino y gemcitabina, seguido de penpulimab, o placebo con cisplatino o carboplatino y gemcitabina, seguido de placebo. La principal variable de eficacia fue la supervivencia libre de progresión (SLP: 9,6 vs 7,0 meses), con un 31 % y un 11 % de pacientes vivos y sin progresión tras 12 meses de seguimiento. La eficacia de penpulimab como agente único se evaluó en un ensayo abierto, multicéntrico y de un solo grupo, realizado en un solo país, que incluyó a 125 pacientes con carcinoma nasofaríngeo no queratinizante irresecable o metastásico con progresión de la enfermedad tras quimioterapia con platino y al menos otra línea de tratamiento. Los pacientes recibieron penpulimab hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable, durante un máximo de 24 meses. La principal variable de eficacia fue la tasa de respuesta objetiva (TRO: 28%).
Eventos adversos: Los más comunes (≥20%) para penpulimab con cisplatino o carboplatino y gemcitabina son náuseas, vómitos, hipotiroidismo, estreñimiento, disminución del apetito, pérdida de peso, tos, infección por COVID-19, fatiga, erupción cutánea y pirexia. Las más frecuentes (≥20%) para penpulimab como agente único son hipotiroidismo y dolor musculoesquelético. Se produjeron reacciones adversas mortales en el 1% de los pacientes, incluyendo un caso de neumonitis, choque séptico, colitis y hepatitis.
LINVOSELTAMAB (LYNOZYFIC®) REGENERON (UE)
Indicación: Tratamiento en monoterapia de pacientes adultos con mieloma múltiple en recaída y refractario que han recibido al menos 3 tratamientos previos, incluidos un inhibidor del proteasoma, un agente inmunomodulador y un anticuerpo monoclonal anti-CD38, y han presentado progresión de la enfermedad al último tratamiento.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal biespecífico recombinante humano de tipo inmunoglobulina (Ig)G4. Autorizado en la Unión Europea el 23 de abril de 2025 condicionalmente (Conditional marketing authorisation); no disponible en Estados Unidos.
Mecanismo: Anticuerpo humano biespecífico de tipo IgG4 que se une al grupo de diferenciación 3 (CD3), un antígeno de los linfocitos T asociado con el complejo receptor de linfocitos T, y al antígeno de maduración de linfocitos B (BCMA), el cual se expresa en la superficie de las células malignas del linaje B del mieloma múltiple, así como en los linfocitos B en la última etapa de maduración y en las células plasmáticas. La interacción simultánea con ambas ramas de linvoseltamab da lugar a la formación de una sinapsis entre el linfocito T y la célula que expresa BCMA, que a su vez produce la activación de los linfocitos T y la generación de la respuesta de linfocitos T citotóxicos policlonales, que da como resultado la lisis redirigida de las células diana, incluidas las células malignas del linaje B del mieloma múltiple.
Eficacia clínica: Estudio en fase I/II abierto, multicéntrico y de cohortes múltiples en pacientes (12 en la fase I y 105 en la fase II) con mieloma múltiple en recaída o refractario que habían recibido al menos 3 tratamientos previos, incluido un inhibidor del proteasoma (IP), un agente inmunomodulador (IMiD) y un anticuerpo anti-CD38. La variable principal de eficacia fue la tasa de respuesta objetiva (TRO) determinada por el comité de revisión independiente: Respuesta completa o mejor (RC: 50%); respuesta parcial (RP: 22%). Las variables secundarias fueron, entre otros, la duración de la respuesta (DR: mediana de 29 meses) y la tasa de negatividad de enfermedad mínima residual en pacientes con RC (EMR: 41%). La mediana del tiempo hasta la respuesta completa o mejor fue de 8 meses.
Eventos adversos: Los más comunes son dolor musculoesquelético (52%), síndrome de liberación de citocinas (46%), neutropenia (43%), tos (42%), diarrea (39%), anemia (38%), fatiga (36%), neumonía (32%) e infección del tracto respiratorio alto (30%). Se produjeron reacciones adversas graves en el 75 % de los pacientes, especialmente síndrome de liberación de citocinas (27%), neumonía (13%), COVID-19 (7%) y lesión renal aguda (5%). El 19% de los pacientes interrumpieron permanentemente el tratamiento debido a reacciones adversas, debido principalmente a neumonía por COVID-19 (1,7%), neumonía por Pneumocystis jirovecii (1,7%) y sepsis por Pseudomonas (1,7%).
NIPOCALIMAB (IMAAVY®) JANSSEN (USA)
Indicación: Tratamiento de la miastenia grave generalizada (MGg) en pacientes adultos y pediátricos de 12 años de edad o mayores que presenten anticuerpos antirreceptor de acetilcolina (AChR) o antitirosina cinasa específica del músculo (MuSK).
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal humano IgG1 aglicosilado. Autorizado en Estados Unidos el 29 de abril de 2025 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no disponible en la Unión Europea.
Mecanismo: Anticuerpo monoclonal dirigido contra el receptor de fragmentos cristalizables neonatales (FcRn), con potencial actividad inmunomoduladora. Nipocalimab se une al FcRn en el sitio de unión de la inmunoglobulina G (IgG), impidiendo así la interacción entre el FcRn y la proteína sérica IgG. Al impedir la unión de FcRn/IgG, nipocalimab bloquea el rescate de IgG mediado por FcRn, permite su degradación y previene la inflamación mediada por IgG. La IgG desempeña un papel clave en numerosas enfermedades autoinmunes y es un factor importante en los procesos inflamatorios.
Eficacia clínica: Estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo de 24 semanas de duración en 196 pacientes. La variable principal de eficacia fue la comparación del cambio medio entre el inicio y las semanas 22, 23 y 24 en la puntuación total de la escala MG-ADL (Miastenia Grave – Actividades de la Vida Diaria) entre los grupos de tratamiento: -4,7 vs -3-3. La variable secundaria consistió en la comparación del cambio medio entre el inicio y las semanas 22 y 24 en la puntuación total de la escala QMG (Miastenia Grave Cuantitativa) entre los grupos de tratamiento: -4,9 vs -2,1.
Eventos adversos: Los más comunes (≥ 5%) y con mayor frecuencia que en placebo son infección de las vías respiratorias (18% vs 13% con placebo), infección de las vías urinarias (6/3%), herpes zóster y herpes simple (6/2%), infección oral (5/3%), edema periférico (12/2%), espasmo muscular (12/3%), reacción de hipersensibilidad (8/7%), dolor abdominal (8/3%), dolor de espalda (8/5%), pirexia (7/1%), diarrea (7/3%), tos (7/3%), anemia (6/4%), mareos (5/1%), náuseas (5/2%), hipertensión (5/2%) e insomnio (5/2%).
AVUTOMETINIB + DEFACTINIB (AVMAPKI FAKZYNJA CO-PACK®) VERASTEM (USA)
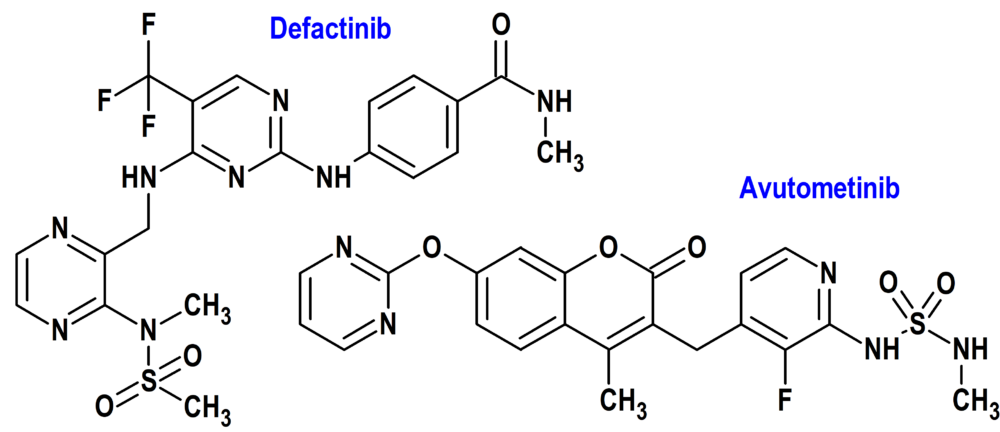
Indicación: Tratamiento de pacientes adultos con cáncer de ovario seroso de bajo grado recurrente (LGSOC) con mutación KRAS que hayan recibido terapia sistémica previa.
Tipo: Medicamento sintético estándar constituido por una combinación de dos inhibidores de tirosina cinasa, avutometinib (N-[3-fluoro-4-({4-metil-2-oxo-7-[(pirimidin-2-il)oxi]-2H1-benzopiran-3-il}metil)piridin-2-il]-N’-metildiamida sulfúrica); y defactinib (N-metil-4-[[4-[[3-[metil(metilsulfonil)amino]pirazin-2-il]metilamino]-5-(trifluorometil)pirimidin-2-il]amino]benzamida). Autorizado en Estados Unidos el 8 de mayo de 2025 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no disponible en la Unión Europea.
Mecanismo: Combinación de dos inhibidores de tirosina cinasa. Avutometinib es un inhibidor de la proteína cinasa activada por mitógenos (MEK); inhibe la proliferación de líneas celulares mutantes BRAFV600E y KRAS. Defactinib es un inhibidor de la cinasa de adhesión focal (FAK), con actividades antiangiogénicas y antineoplásicas.
Eficacia clínica: Un ensayo multicéntrico abierto que incluyó a 57 pacientes adultos con LGSOC recurrente con mutación medible en KRAS. Los pacientes debían haber recibido al menos un tratamiento sistémico previo, incluyendo un régimen basado en platino. La variable principal de eficacia fue la tasa de respuesta global (TRO: 44%), evaluada por un comité de revisión independiente. Otra variable de eficacia fue la duración de la respuesta (DR: rango de 3,3 a 31,1 meses).
Eventos adversos: Los más comunes (≥25%) son aumento de la creatina fosfoquinasa, náuseas, fatiga, aumento de la aspartato aminotransferasa, erupción cutánea, diarrea, dolor musculoesquelético, edema, disminución de la hemoglobina, aumento de la alanina aminotransferasa, vómitos, aumento de la bilirrubina en sangre, aumento de los triglicéridos, disminución del recuento de linfocitos, dolor abdominal, dispepsia, dermatitis acneiforme, trastornos vítreo-retinianos, aumento de la fosfatasa alcalina, estomatitis, prurito, deterioro visual, disminución del recuento de plaquetas, estreñimiento, piel seca, disnea, tos, infección del tracto urinario y disminución del recuento de neutrófilos.
TELISOTUZUMAB VEDOTINA (EMRELIS®) ABBVIE (USA)

Indicación: Tratamiento de adultos con cáncer de pulmón de células no pequeñas (CPCNP) no escamoso, localmente avanzado o metastásico, con alta sobreexpresión de la proteína c-Met [≥50 % de células tumorales con tinción fuerte (3+)], según lo determinado por una prueba aprobada por la FDA, que hayan recibido una terapia sistémica previa.
Tipo: Medicamento biológico constituido por un anticuerpo humano conjugado con vedotina, un agente disruptor de microtúbulos unido al anticuerpo a través de un enlazador escindible por proteasa. Autorizado en Estados Unidos el 14 de mayo de 2025 mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no disponible en la Unión Europea.
Mecanismo: Telisotuzumab es un anticuerpo bivalente recombinante humano contra el receptor del factor de crecimiento de hepatocitos (HGFR, también denominado c-MET o tirosina-proteína cinasa Met; una proteína que en los seres humanos está codificada por el gen MET), al que se une con alta afinidad e inhibe la señalización de c-Met. La vedotina o monometil auristatina E (MMAE) es un agente disruptor de microtúbulos, que se encuentra unido al anticuerpo a través de un enlazador escindible por proteasa, tras lo que ejerce una acción tóxica en el interior de las células sobre las que se fija selectivamente el telisotuzumab.
Eficacia clínica: Ensayo multicéntrico, abierto y multicohorte, que incluyó a 84 pacientes con CPNM no escamoso con receptor del factor de crecimiento epidérmico (EGFR) de tipo salvaje y alta sobreexpresión de la proteína c-Met, que habían recibido terapia sistémica previa. Las principales variables de eficacia fueron la tasa de respuesta global (TRO: 35%) y la duración de la respuesta (DDR: mediana de 7,2 meses), determinadas mediante una revisión central independiente y enmascarada (BICR) según RECIST 1.1.
Eventos adversos: Los más comunes (≥20%) son neuropatía periférica, fatiga, disminución del apetito y edema periférico. Las anomalías de laboratorio de grado 3 o 4 más frecuentes (≥2%) fueron disminución de linfocitos, aumento de glucosa, aumento de alanina aminotransferasa, aumento de gamma glutamil transferasa, disminución de fósforo, disminución de sodio, disminución de hemoglobina y disminución de calcio.
TALETRACTINIB (IBTROZI®) NUVATION (USA)

Indicación: Tratamiento de pacientes adultos con cáncer de pulmón de células no pequeñas (CPCNP) localmente avanzado o metastásico con ROS1 positivo.
Tipo: Medicamento sintético estándar constituido por la 3-{4-[(2R)-2-aminopropoxi]fenil}-N-[(1R)-1-(3-fluorofenil)etil]imidazo [1,2-b]piridazin-6-amina. Autorizado en Estados Unidos el 11 de junio de 2025 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review); no disponible en la Unión Europea.
Mecanismo: Inhibidor de la tirosina cinasa ROS1, incluyendo mutaciones de resistencia a ROS1. Taletrectinib también tiene efectos inhibidores sobre las cinasas del receptor de tropomiosina (TRK) TRKA, TRKB y TRKC. Las proteínas de fusión que incluyen dominios ROS1 pueden impulsar el potencial tumorigénico mediante la hiperactivación de las vías de señalización posteriores, lo que conduce a una proliferación celular sin restricciones. Taletrectinib inhibe el crecimiento de células cancerosas que expresan genes de fusión y mutaciones de ROS1.
Eficacia clínica: Dos ensayos clínicos multicéntricos, de un solo brazo y abiertos realizados con 157 pacientes (103+54) sin tratamiento previo con un inhibidor de la tirosina cinasa (ITK) de ROS1 y a 113 (66+47) que habían sí lo habían recibido previamente; los pacientes podrían haber recibido quimioterapia previa para la enfermedad avanzada. Las variables principales de eficacia fueron la tasa de respuesta global (TRO) confirmada y la duración de la respuesta (DDR), determinadas mediante una revisión central independiente y enmascarada. En pacientes sin tratamiento previo, la TRO fue del 90% en el primer estudio y del 85% en el segundo, con una DDR ≥12 meses en el 72 % y el 63 % de los respondedores, respectivamente. En pacientes pretratados con TKI, la TRO fue del 52% y 62%, con una DDR ≥6 meses en el 74 % y el 83 % de los respondedores, respectivamente.
Eventos adversos: Los más comunes (≥20%) son diarrea, náuseas, vómitos, mareos, erupción cutánea, estreñimiento y fatiga. Las anomalías de laboratorio de grado 3 o 4 más frecuentes (≥2%) fueron aumento de ALT, aumento de AST, disminución de neutrófilos, aumento de creatinfosfocinasa, disminución de linfocitos, aumento de magnesio, disminución de hemoglobina y aumento de triglicéridos. El 31% de los pacientes presentó reacciones adversas graves, incluyendo neumonía (7%), derrame pleural (4,7%) y hepatotoxicidad (2,4%). El 5% presentó reacciones adversas mortales, incluyendo neumonía (2,4%), síndrome de disfunción multiorgánica (0,6%), hepatotoxicidad (0,6%), paro cardíaco (0,6%), insuficiencia cardíaca (0,3%), insuficiencia cardiopulmonar (0,3%) e insuficiencia respiratoria (0,3%). El 7% de los pacientes requirió la suspensión definitiva del tratamiento, incluyendo neumonía, enfermedad pulmonar intersticial (EPI) y hepatotoxicidad. En el 41% de los pacientes se produjeron interrupciones de la dosis debido a una reacción adversa, incluido un aumento de AST y de ALT.
TEPROTUMUMAB (TEPEZZA®) AMGEN (UE; USA)
Indicación: Tratamiento de adultos con enfermedad ocular tiroidea (EOT) de moderada a grave.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal IgG1 completamente humano. Autorizado en la Unión Europea el 19 de junio de 2025; disponible previamente en Estados Unidos desde el 21 de enero de 2020.
Mecanismo: Inhibidor del receptor del factor de crecimiento similar a la insulina 1 (IGF-1R), bloqueando su activación y señalización. El mecanismo de acción en pacientes con enfermedad ocular tiroidea no ha sido completamente caracterizado, aunque podría bloquear la activación autoinmune de los fibroblastos orbitarios, inhibiendo potencialmente el desarrollo y la progresión de la enfermedad ocular tiroidea (enfermedad ocular de Graves).
Eficacia clínica: Tres ensayos aleatorizados y controlados con placebo en un total de 225 pacientes con EOT activa y un ensayo en 62 pacientes con EOT crónica. Tras 24 semanas, los pacientes tratados con Tepezza experimentaron una reducción significativa (de -2 a -2,3 mm) de la protrusión del globo ocular respecto a la órbita (proptosis) y de la Escala de Actividad Clínica (CAS), una herramienta estándar para evaluar los signos y síntomas inflamatorios de la EOT, en comparación con los pacientes tratados con placebo. La reducción de la proptosis fue menor (-1,5 mm) en los pacientes con EOT crónica.
Eventos adversos: Los más comunes son espasmos musculares, diarrea, alopecia, hiperglucemia, fatiga, náuseas y cefalea. Puede causar pérdida auditiva grave, incluyendo pérdida de audición, que en algunos casos puede ser permanente. Estudios preclínicos han demostrado que el medicamento puede suponer riesgos para el desarrollo fetal.
SARGRAMOSTIM (IMREPLYS®) PARTNER (UE; USA)
Indicación: Tratamiento de personas con síndrome hematopoyético de radiación aguda (SRA-H) tras la exposición aguda a dosis mielosupresoras de radiación.
Tipo: Medicamento biológico constituido por una forma recombinante del factor estimulante de colonias de granulocitos y macrófagos (GM-CSF) humano; se trata de una glicoproteína de 127 aminoácidos, cuya secuencia difiere del GM-CSF humano natural por una sustitución de leucina en la posición 23, donde la leucina sustituye a la arginina, y la fracción de carbohidrato puede ser diferente a la de la proteína nativa. Evaluado favorablemente en la Unión Europea (CHMP, EMA) el 19 de junio de 2025 en condiciones excepcionales (Exceptional circumstances); en Estados Unidos está comercializado como LEUKINE®, aunque no para la misma indicación que la autorizada ahora en la Unión Europea.
Mecanismo: El sargramostim (GM-CSF) pertenece a un grupo de factores de crecimiento denominados factores estimulantes de colonias (CEC), que favorecen la supervivencia, la expansión clonal y la diferenciación de las células progenitoras hematopoyéticas. El GM-CSF induce la división y diferenciación de las células progenitoras parcialmente comprometidas en las vías granulocito-macrófago, que incluyen neutrófilos, monocitos/macrófagos y células dendríticas de origen mieloide. El GM-CSF también es capaz de activar granulocitos y macrófagos maduros. Es un factor multilinaje y, además de sus efectos dosis-dependientes sobre el linaje mielomonocítico, puede promover la proliferación de progenitores megacariocíticos y eritroides. Sin embargo, se requieren otros factores para inducir la maduración completa en estos dos linajes. Las diversas respuestas celulares (división, maduración y activación) se inducen mediante la unión del GM-CSF a receptores específicos expresados en la superficie celular de las células diana.
Eficacia clínica: Los beneficios clínicos en humanos han sido extrapolados a partir de la consecución de mayores tasas de supervivencia a los 60 días en monos Rhesus que recibieron irradiación corporal total inductora de H-ARS, en comparación con placebo, como se muestra en 3 estudios aleatorizados, ciegos y controlados con placebo. Todos los estudios también han demostrado una recuperación más rápida de los recuentos absolutos de neutrófilos y plaquetas, tasas de infección reducidas y menos signos de sepsis.
Eventos adversos: Los más comunes son fiebre, diarrea, vómitos, reacciones cutáneas, erupción cutánea, astenia, anomalías metabólicas de laboratorio, malestar general, niveles altos de glucosa, dolor abdominal, pérdida de peso, niveles bajos de albúmina, prurito, hemorragia gastrointestinal, escalofríos, faringitis, dolor óseo, dolor torácico, hipomagnesemia, hematemesis, artralgia, ansiedad y hemorragia ocular.
(N) SISTEMA NERVIOSO
DIFLUNISAL (ATTROGY®) PURPOSE (UE)

Indicación: Tratamiento de la amiloidosis hereditaria mediada por transtiretina (amiloidosis hATTR) en pacientes adultos con polineuropatía en estadio 1 o estadio 2.
Tipo: Medicamento sintético estándar constituido por el ácido 5-(2,4-difluorofenil)-2-hidroxibenzoico. Evaluado favorablemente en la Unión Europea (CHMP, EMA) el 25 de abril de 2025 como medicamento huérfano (Orphan drug). Inicialmente comercializado en Europa y Estados Unidos en la década de 1980, actualmente ya no lo está en la Unión Europea, mientras que en Estados Unidos está presente en varios medicamentos genéricos como analgésico convencional pero no con la indicación ahora autorizada en la UE.
Mecanismo: El diflunisal estabiliza el tetrámero de transtiretina (TTR), impidiendo su disociación en monómeros de TTR, responsables de la amiloidosis por TTR.
Eficacia clínica: Ensayo aleatorizado, doble ciego y controlado con placebo. Los beneficios de Attrogy residen en su capacidad para retrasar la progresión de la enfermedad, produciendo una mejora de dos a tres veces – en comparación con placebo – en las puntuaciones de deterioro de la neuropatía, medida mediante la Escala de Deterioro Neuropático más 7 pruebas nerviosas (NIS+7), tras 2 años de tratamiento.
Eventos adversos: Los más comunes son empeoramiento de función renal, epigastralgia y hemorragia menor, generalmente de carácter leve y ocurren en los primeros meses de tratamiento.
(S) ÓRGANOS SENSORIALES
REVAKINAGENE TARORETCEL (ENCELTO®) NEUROTECH (USA)
Indicación: Tratamiento de adultos con telangiectasia macular idiopática tipo 2 (MacTel).
Tipo: Medicamento de terapia avanzada (génica), constituido por células encapsuladas alogénicas que contiene de 200.000 a 440.000 células epiteliales pigmentarias de la retina alogénicas que expresan el factor neurotrópico ciliar humano recombinante (rhCNTF) (línea celular NTC-201-6A), para su colocación intravítrea quirúrgica. Autorizado (FDA) en Estados Unidos el 5 de marzo de 2025 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval) y mediante revisión prioritaria (Priority Review); no disponible en la Unión Europea.
Mecanismo: Secreta factor neurotrófico ciliar humano recombinante (rhCNTF), uno de varios factores neurotróficos producidos endógenamente por neuronas y células gliales de soporte. Se cree que el CNTF exógeno actúa inicialmente sobre la glía de Müller para desencadenar una cascada de eventos de señalización que podrían promover la supervivencia de los fotorreceptores. El tipo 2 es la forma más común de telangiectasia macular, en que los vasos sanguíneos se dilatan y presentan fugas, lo que causa inflamación de la mácula . En ocasiones, se pueden formar nuevos vasos sanguíneos debajo de la retina y también romperse o presentar fugas. Con el tiempo, puede producirse cicatrización sobre la mácula, lo que afecta gravemente la visión.
Eficacia clínica: Dos ensayos clínicos aleatorizados, multicéntricos y controlados con placebo en pacientes adultos con MacTel (115 y 113 pacientes, respectivamente). En ambos estudios la variable principal de eficacia fue la tasa de cambio en el área de pérdida de la zona elipsoide (ZE) a lo largo de 24 meses, medida mediante tomografía de coherencia óptica de dominio espectral (SD-OCT): 0,75 vs 0,166 mm² (estudio 1) y 0,111 vs 0,160 mm², mientras que la variable secundaria fue el cambio medio en la pérdida de sensibilidad agregada de la microperimetría dentro del área de ruptura de la ZE desde el inicio hasta el mes 24: 25,27 vs 43,01 dB en el estudio 1 y 40,02 vs 41,97 dB en el estudio 2.
Eventos adversos: Los más comunes (≥2%) son fueron hemorragia conjuntival, retraso en la adaptación a la oscuridad, sensación de cuerpo extraño, dolor ocular, complicaciones relacionadas con la sutura, miosis, hiperemia conjuntival, prurito ocular, molestias oculares, hemorragia vítrea, visión borrosa, dolor de cabeza, ojo seco, irritación ocular, progresión o formación de cataratas, miodesopsias, pérdida grave de la visión, secreción ocular, células de la cámara anterior e iridociclitis.
ACOLTREMON (TRYPTYR®) ALCON (USA)
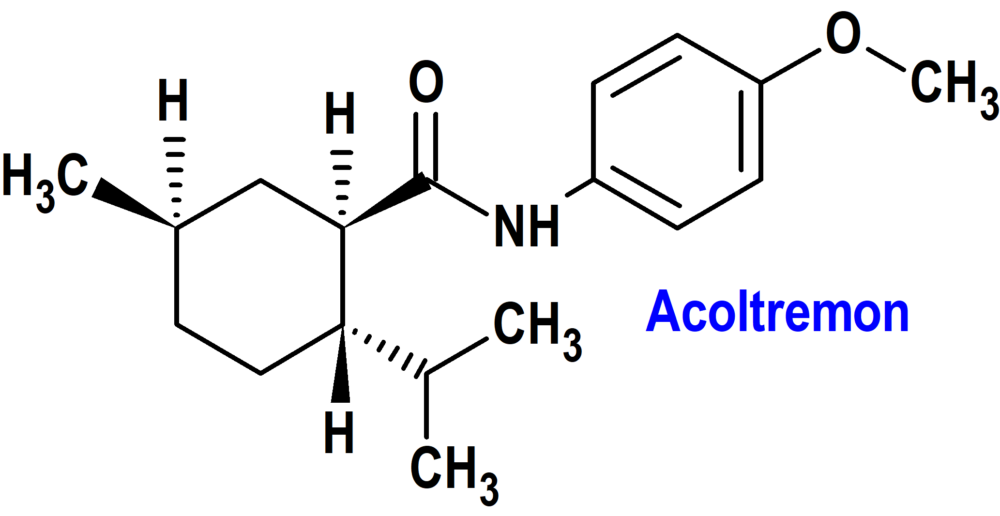
Indicación: Tratamiento de los signos y síntomas de la enfermedad del ojo seco (EOS).
Tipo: Medicamento sintético estándar constituido por la (1R,2S,5R)-5-metil-N-(4-metoxifenil)-2-(propan-2-il)ciclohexano-1-carboxamida . Autorizado (FDA) en Estados Unidos el 28 de mayo de 2025; no disponible en la Unión Europea.
Mecanismo: Agonista selectivo del canal de calcio TRPM8 (Miembro 8 de la subfamilia M del canal catiónico Potencial del Receptor Transitorio), responsable de la sensación de frío producida por el mentol. Se ha demostrado que la estimulación del termorreceptor TRPM8 activa la señalización del nervio trigémino, lo que aumenta la producción basal de lágrimas.
Eficacia clínica: Dos ensayos clínicos de fase 3 con más de 930 pacientes (aleatorizados y controlados con placebo: vehículo) con antecedentes de EOS y una edad promedio de 61 años (30-93), mayoritariamente mujeres (75%); los criterios de inclusión incluyeron signos (puntuación de tinción corneal con fluoresceína [2-15] y prueba de Schirmer lagrimal con anestesia [2-9 mm]) y síntomas (puntuación SANDE [≥50] y puntuación de molestias oculares [≥50]) de la enfermedad del ojo seco. La producción de la película lagrimal se midió mediante la prueba de Schirmer lagrimal sin anestesia, evaluada con una tira de Schirmer (0-35 mm). Las puntuaciones de Schirmer basales promedio sin anestesia para los pacientes tratados con acoltremon y el vehículo fueron de 6,2 mm y 5,9 mm en el primer estudio, y de 6,8 mm y 6,4 mm en el segundo.Los porcentajes de pacientes que recibieron acoltremon y experimentaron un aumento de al menos 10 mm en la producción natural de lágrimas el día 14, en comparación con el placebo, fueron 42,6 vs 8,2% en el primer estudio y 53,2 vs 14,4% en el segundo. Se observaron resultados consistentes en todos los puntos temporales hasta el día 90, observándose que la producción natural de lágrimas en los pacientes tratados con acoltremon fue estadísticamente significativa desde el día 1.
Eventos adversos: Los más comunes son dolor en el lugar de instilación (50%). Menos del 1 % de los pacientes interrumpieron el tratamiento debido a una sensación de ardor o escozor ocular.
PROCEDIMIENTOS ESPECIALES DE EVALUACIÓN Y AUTORIZACIÓN
Tanto la Agencia Europea de Medicamentos (European Medicines Agency, EMA), de la Unión Europea, como la Administración de Alimentos y Medicamentos (Food & Drug Administration, FDA), de Estados Unidos, disponen de diversos procedimientos de evaluación y autorización de medicamentos para incentivar el desarrollo de nuevos tratamientos para enfermedades que de otra manera no atraerían el interés de las empresas debido al elevado coste del desarrollo y la imposibilidad de retorno económico comercial, así como para facilitar la mejor y más rápida disponibilidad posible de medicamentos designados como especialmente relevantes atendiendo a las particulares características patológicas de algunos pacientes, así como a la gravedad de las patologías para los que son destinados y a su potencial repercusión social y epidemiológica, valorando si constituyen el primer tratamiento disponible o si presentan ventajas significativas sobre los tratamientos existentes. Estas designaciones y procedimientos son referenciados, en su caso, en las monografías de los medicamentos previamente descritas.
EMA (European Medicines Agency, UE)
El dictamen favorable adoptado por el CHMP (Committee for Medicinal Products for Human Use) es un paso imprescindible pero no definitivo en el proceso de acceso del medicamento a los pacientes. Ese dictamen es enviado a la Comisión Europea (CE), que es la autoridad máxima responsable de la AUTORIZACIÓN de comercialización del medicamento para el conjunto de los Estados miembros de la Unión Europea (un proceso que, salvo situaciones de emergencia, dura 60 días o más, según las circunstancias); sin embargo, las decisiones sobre el precio, la financiación pública y, en su caso, el tipo de reembolso se toman en cada Estado miembro, teniendo en cuenta el posible papel o uso de este medicamento en el contexto del sistema nacional de salud de dicho país.
Medicamentos Prioritarios (Priority Medicines; PRIME): es un esquema de evaluación de la EMA para apoyar el desarrollo de medicamentos que se dirigen a una necesidad médica no cubierta, basándose en una interacción mejorada y un diálogo temprano con los desarrolladores de medicamentos prometedores, para optimizar los planes de desarrollo y acelerar la evaluación para que estos medicamentos puedan llegar antes a los pacientes, empleando para ello el asesoramiento científico y la evaluación acelerada.
Evaluación acelerada (Accelerated assessment): reduce el plazo máximo para que el Comité de Medicamentos de Uso Humano (CHMP) revise una solicitud de autorización de comercialización de medicamentos, pasando de 210 a 150 días. Las solicitudes pueden ser elegibles para una evaluación acelerada si el CHMP decide que el producto es de gran interés para la salud pública y la innovación terapéutica.
Autorización de comercialización condicional (Conditional marketing authorisation) para solicitudes de medicamentos que presenten datos clínicos menos completos que los normalmente requeridos, siempre que el beneficio de la disponibilidad inmediata del medicamento supere el riesgo inherente al hecho de que todavía se requieren datos adicionales, tal como aquellos destinados a tratar, prevenir o diagnosticar enfermedades gravemente debilitantes o potencialmente mortales, incluyendo a los medicamentos huérfanos.
Autorización de comercialización en condiciones excepcionales (Exceptional circumstances) para medicamentos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y la seguridad en condiciones normales de uso, porque la condición a tratar es rara o porque la recopilación de información completa no es posible o no es ético.
Medicamento huérfano (Orphan drug): son designados como tales aquellos destinados a tratar enfermedades raras (en la Unión Europea son aquellas que afectan a menos de 5 de cada 10.000 habitantes), no resultan atractivos a los patrocinadores por su escasa rentabilidad y precisan por ello apoyo adicional para su desarrollo.
FDA (Food & Drug Administration, USA)
Revisión prioritaria (Priority Review): evaluación de solicitudes de medicamentos que, de aprobarse, serían mejoras significativas en la seguridad o eficacia del tratamiento, diagnóstico o prevención de afecciones graves en comparación con las solicitudes estándar, considerando mejora significativa a la evidencia de mayor efectividad en el tratamiento, prevención o diagnóstico de la condición; eliminación o reducción sustancial de una reacción farmacológica limitante del tratamiento; mejora documentada del cumplimiento del paciente que se espera que conduzca a una mejora en los resultados graves; o evidencia de seguridad y eficacia en una nueva subpoblación.
Bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD): la FDA puede otorgar bonos o cupones de revisión prioritaria a los patrocinadores de aplicaciones de productos destinados para enfermedades pediátricas raras que cumplan con ciertos criterios. Este bono es un incentivo que el patrocinador recibe en forma de “cupón especial”, el cual puede ser empleado de dos maneras: para aplicar el sistema de revisión prioritaria de la FDA en cualquier otro de sus productos o venderlo a otra compañía interesada en que su propio medicamento sea revisado de forma prioritaria.
Terapia innovadora (Breakthrough Therapy): medicamentos destinados a tratar una afección grave y cuya evidencia clínica preliminar indica que puede demostrar una mejora sustancial sobre la terapia disponible en una o varias variables clínicamente significativas, como la duración del efecto, la relevancia del resultado clínico observado mostrando una clara ventaja sobre la terapia disponible.
Autorización acelerada (Accelerated Approval): medicamentos indicados en afecciones graves que cubran una necesidad médica no satisfecha, que puedan ser autorizados precozmente basándose en una a más variables subrogadas (una medida de laboratorio o signo físico que se usa como sustituto de una variable clínicamente significativa que es una medida directa sobre lo que siente un paciente, sus funciones o su supervivencia y que se espera que prediga el efecto de la terapia).
Vía rápida (Fast Track): medicamentos que aborden enfermedades graves en las que puedan tener un impacto significativo sobre la supervivencia, el funcionamiento diario o la probabilidad de que la afección, si no se trata, progrese de una condición menos severa a una más severa, tales como el SIDA, la enfermedad de Alzheimer, la insuficiencia cardíaca y o cáncer.
Medicamento huérfano (Orphan drug): designación de un medicamento potencialmente útil para prevenir, diagnosticar o tratar una enfermedad rara; es decir, con menos de 200.000 pacientes/año (los que supone una prevalencia aproximada de 7,5/10.000 habitantes, en la actualidad).
Terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy): cualquier medicamento de terapia celular, de ingeniería tisular, de células y tejidos humanos, o cualquier combinación de dichas terapias o productos, que esté destinado a tratar, modificar, revertir o curar una enfermedad o afección grave o potencialmente mortal; y que la evidencia clínica preliminar indica que el medicamento tiene el potencial de abordar necesidades médicas no cubiertas para dicha enfermedad o afección.
Producto Calificado para Enfermedades Infecciosas. (Qualified Infectious Disease Product): un medicamento antibacteriano o antifúngico para uso humano destinado a tratar infecciones graves o potencialmente mortales, incluidas aquellas causadas por un patógeno resistente a antibacterianos o antifúngicos, incluidos patógenos infecciosos nuevos o emergentes.